blast
使用建议
module 中配好了NR、NT等公共数据库,module load之后按提示使用,可不用自己下载
使用diomand可替代blastx blastp,可以极大地提高比对速度,module中有格式化好的nr库可直接使用
blastn 如果运行比较慢,建议将query序列分割多个序列,提交多个作业同时跑,可以提高跑的速度,因为blastn的CPU利用率不高,线程数建议设置4或8左右即可
BLAST+ 2.15.0 速度有所改善,见 https://www.ncbi.nlm.nih.gov/books/NBK131777/
基本使用¶
构建索引¶
$ module load BLAST/2.15.0
# 对核酸序列构建索引
$ makeblastdb -in ref.fa -dbtype nucl -parse_seqids
# 对氨基酸序列构建索引
$ makeblastdb -in prot.fa -dbtype prot -parse_seqids
# 生成的索引文件
ref.fa
ref.fa.nhr
ref.fa.nin
ref.fa.nog
ref.fa.nsd
ref.fa.nsi
ref.fa.nsq
blastn¶
一般用法
$ blastn -db ref.fa -outfmt 6 -num_threads 6 -evalue 0.001 -query query.fa -out blast_out
如果 ref.fa 序列比较小,可以不用构建索引,使用 -subject 选项直接比对。
$ blastn -subject ref.fa -outfmt 6 -num_threads 6 -query query.fa -out blast_out
超短序列比对¶
blastn 的query序列默认需要大于50nt,对于query小于50nt的短序列,可使用 -task blastn-short 选项并配合其他选项使用,否则可能没有结果出来。
$ blastn -task blastn-short -outfmt 6 -num_threads 6 -word_size 4 -gapopen 1 -gapextend 1 -db ref.fa -query query.fa -out blast_out
输出格式¶
-outfmt <String> 支持的输出格式
alignment view options:
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
outfmt 6,其各列的定义为: [00] Query id
[01] Subject id
[02] % identity
[03] alignment length
[04] mismatches
[05] gap openings
[06] q. start
[07] q. end
[08] s. start
[09] s. end
[10] e-value
[11] bit score
格式6, 7, 10 支持自定义输出列。
The supported format specifiers for options 6, 7 and 10 are:
qseqid means Query Seq-id
qgi means Query GI
qacc means Query accession
qaccver means Query accession.version
qlen means Query sequence length
sseqid means Subject Seq-id
sallseqid means All subject Seq-id(s), separated by a ';'
sgi means Subject GI
sallgi means All subject GIs
sacc means Subject accession
saccver means Subject accession.version
sallacc means All subject accessions
slen means Subject sequence length
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Aligned part of query sequence
sseq means Aligned part of subject sequence
evalue means Expect value
bitscore means Bit score
score means Raw score
length means Alignment length
pident means Percentage of identical matches
nident means Number of identical matches
mismatch means Number of mismatches
positive means Number of positive-scoring matches
gapopen means Number of gap openings
gaps means Total number of gaps
ppos means Percentage of positive-scoring matches
frames means Query and subject frames separated by a '/'
qframe means Query frame
sframe means Subject frame
btop means Blast traceback operations (BTOP)
staxid means Subject Taxonomy ID
ssciname means Subject Scientific Name
scomname means Subject Common Name
sblastname means Subject Blast Name
sskingdom means Subject Super Kingdom
staxids means unique Subject Taxonomy ID(s), separated by a ';'
(in numerical order)
sscinames means unique Subject Scientific Name(s), separated by a ';'
scomnames means unique Subject Common Name(s), separated by a ';'
sblastnames means unique Subject Blast Name(s), separated by a ';'
(in alphabetical order)
sskingdoms means unique Subject Super Kingdom(s), separated by a ';'
(in alphabetical order)
stitle means Subject Title
salltitles means All Subject Title(s), separated by a '<>'
sstrand means Subject Strand
qcovs means Query Coverage Per Subject
qcovhsp means Query Coverage Per HSP
qcovus means Query Coverage Per Unique Subject (blastn only)
When not provided, the default value is:
'qaccver saccver pident length mismatch gapopen qstart qend sstart send
evalue bitscore', which is equivalent to the keyword 'std'
The supported format specifier for option 17 is:
SQ means Include Sequence Data
SR means Subject as Reference Seq
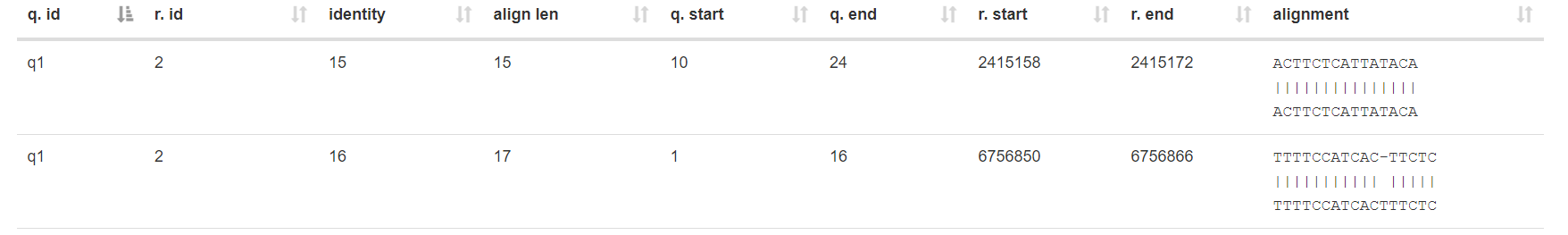
Biopython解析blast结果¶
blast结果中,outfmt 5 生成的XML结果较为丰富,适合用Biopython解析并提取自己想要的结果,需要先安装一下 pip install biopython。
from Bio.Blast import NCBIXML
# 读取 BLAST XML 文件
result_handle = open(blast_output)
blast_records = NCBIXML.parse(result_handle)
data = []
for blast_record in blast_records:
for alignment in blast_record.alignments:
for hsp in alignment.hsps:
align_item = hsp.query + "\n" + hsp.match + "\n" + hsp.sbjct
row_data = {
'col1': blast_record.query,
'col2': alignment.hit_id,
'col4': hsp.identities,
'col5': hsp.align_length,
'col6': hsp.query_start,
'col7': hsp.query_end,
'col8': hsp.sbjct_start,
'col9': hsp.sbjct_end,
'col10': align_item
}
data.append(row_data)
nt_core¶
Interested in faster nucleotide BLAST searches with more focused search results? As previously announced, NCBI has been re-evaluating the BLAST nucleotide database (nt) to make it more compact and more efficient. Thanks to your feedback, NCBI’s BLAST is excited to introduce the core nucleotide database (core_nt), an alternative to the default nt database that contains better-defined content and is less than half the size.
Benefits of BLAST core_nt over nt¶
- Enables faster searches
- Returns similar top results for most searches
- Reduces redundancy for some highly represented organisms
- Allows easier download and requires less storage space for database download for standalone BLAST
What is core_nt?¶
Core_nt contains the same eukaryotic transcript and gene-related sequences as nt. The core_nt database is nt without most eukaryotic chromosome sequences. Most nucleotide BLAST searches with core_nt will be similar to the nt database. However, core_nt is better than nt for accomplishing your most common BLAST search goals, such as identifying gene-related sequences like transcript sequences and complete bacterial chromosomes. This is because, in recent years, nt has acquired more low-relevance, non-annotated, and non-gene content.
参考¶
本文阅读量 次本站总访问量 次