genomicsbench
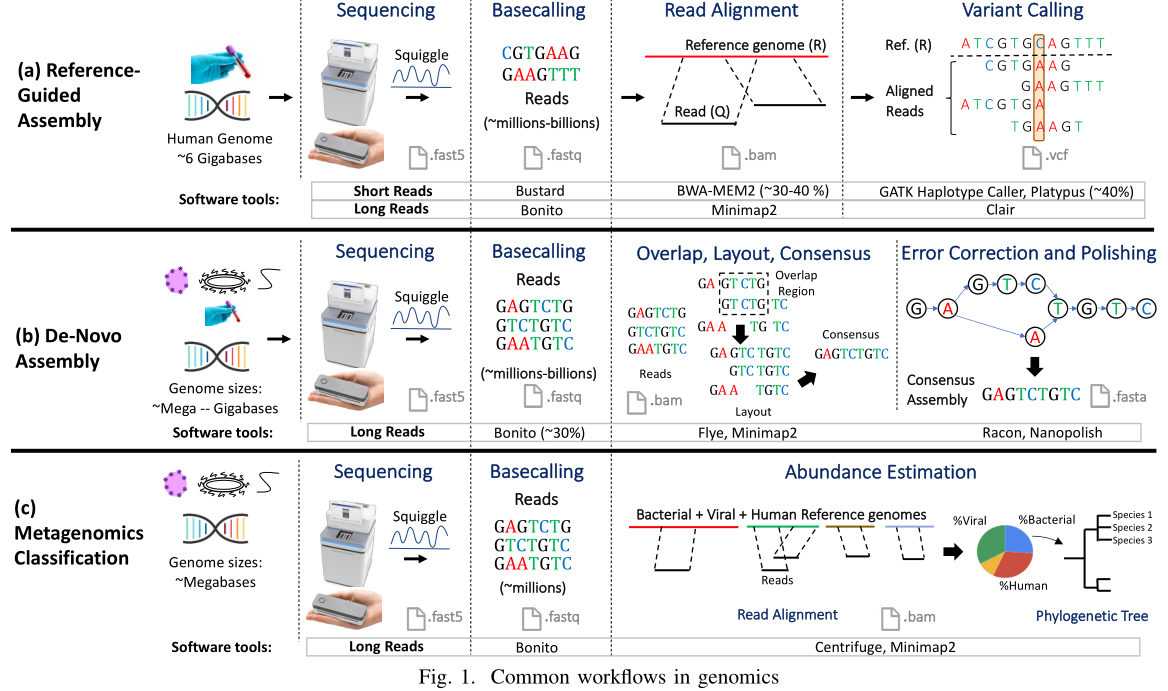
生信软件种类五花八门,每个软件的计算特性不尽相同,很难用某一款软件来做基准测试。文章作者提出了一个基准套件 GenomicsBench,其中包含了12个来自常用生物信息学软件工具的计算密集型数据并行内核,涵盖了短读段和长读段基因组序列分析流水线的主要步骤。
文章地址:GenomicsBench: A Benchmark Suite for Genomics
代码库:https://github.com/arun-sub/genomicsbench
计算核心介绍¶
GenomicsBench基准测试套件包含了12个计算密集型的数据并行核心,以下是这12个计算过程的简要介绍:
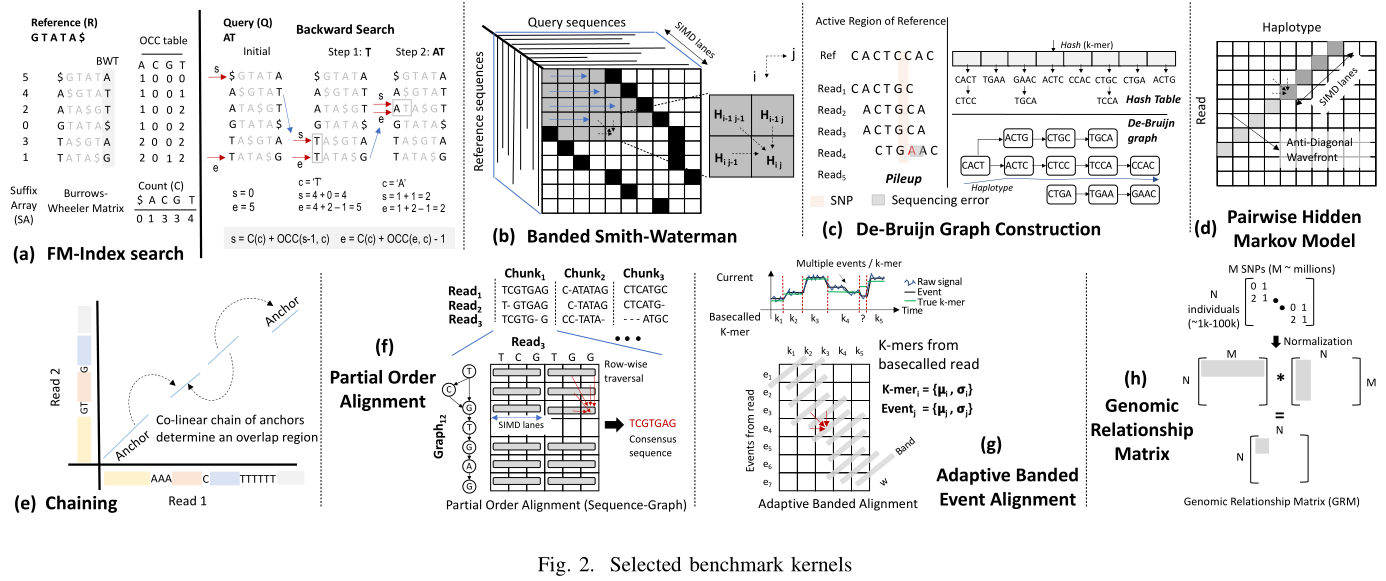
FM-Index Search (fmi): 使用FM-index(Full-Text Index in Minute Space)数据结构来识别参考基因组中与读取序列匹配的子字符串位置。其被用于部分常用比对软件中如BWA、Bowtie2等,用于在参考基因组中识别seeds序列的短匹配子串,特点是低内存占用、能够处理任意长度的子串以及支持不完全匹配。
Banded Smith-Waterman (bsw): Smith-Waterman 是一种动态规划算法,用于估计成对序列之间的相似性,常用于序列比对工具,如BWA-MEM,以及变异调用工具,如GATK HaplotypeCaller,并且该算法是软件中的主要计算瓶颈。
De-Bruijn Graph Construction (dbg): 利用 k-mer 构建 De-Bruijn 图,并在图中寻找表示基因组序列的路径。变异检测工具如GATK HaplotypeCaller和Platypus,通过De-Bruijn图可以识别个体基因组中的小变异(SNPs/indels);大部分二代数据组装软件通过构建 De-Bruijn 图进行基因组组装,代表软件 SOAPdenovo、allpathlg。
Pairwise Hidden Markov Model (pairHMM): pairHMM是一种使用隐马尔可夫模型(HMM)的比对算法。代表软件GATK HaplotypeCaller,其使用隐马尔可夫模型(HMM)对每个reads和每个候选单倍型(haplotype)之间的进行双序列比对(Pairwise alignment),以确定最可能被该reads支持的单倍型。pairHMM与Smith-Waterman的不通点在于,pairHMM主要使用了浮点计算。这里使用了GATK HaplotypeCaller中的pairHMM实现。
Chaining (chain): 使用 long reads 进行 denovo 组装时,估算 reads 之间的 overlap 是最耗时的一步计算过程之一。Chaining 的目标是将一组共线的种子(co-linear seeds)组合成一个重叠区(overlapping region)。Chaining 算法基于一维动态规划,它比较每个种子与之前的一定数量的种子(默认为25)以确定其最佳父节点,从而构建重叠区。
Partial-Order Alignment (poa): poa 是一种用比对算法,用于使用 long reads 对基因组进行 polishing 过程中,代表软件racon。
Adaptive Banded Signal to Event Alignment (abea): abea 是一种用于处理ont数据的比对算法,主要应用在使用ont数据对组装好的基因组进行抛光,以及甲基化位点检测,代表软件 Nanopolish。处理的数据为ont的数据,主要使用GPU计算。
Genomic Relationship Matrix (grm): 大规模的群体遗传学中,通过计算一个N×N的矩阵(基因组关系矩阵,GRM),来计算所有个体之间的潜在祖先关系( potential ancestral relationship)。计算过程主要为浮点计算,对内存带宽要求较高。代表软件plink2。
Neural Network-based Base Calling (nn-base): 在纳米孔基因组测序中,将原始的纳米孔信号数据转换为核苷酸序列的过程称为碱基识别(Base Calling),一般基于神经网络来实现,代表软件Bonito。处理的数据为ont的数据,主要使用GPU计算。
Pileup Counting (pileup): 在长读取神经网络变异调用工具中,如 Medaka,涉及解析比对数据以生成不同基因组位置的读段堆叠(read pileup),并计算不同碱基、插入和删除的计数。处理的数据为ont的数据,Medaka可以使用CPU和GPU计算。
Neural Network-based Variant Calling (nn-variant): long reads 变异检测工具检查特定基因组参考位置的读段堆叠,并检测与该参考基因组同源和杂合的变异。处理的数据为ont的数据,主要使用GPU计算。
K-mer Counting (kmer-cnt): K-mer计数是基因组学中一个常见的任务,用于计数输入读取中每个唯一k-mer的出现次数,文章中选用的Flye中的 kmer-cnt 实现。
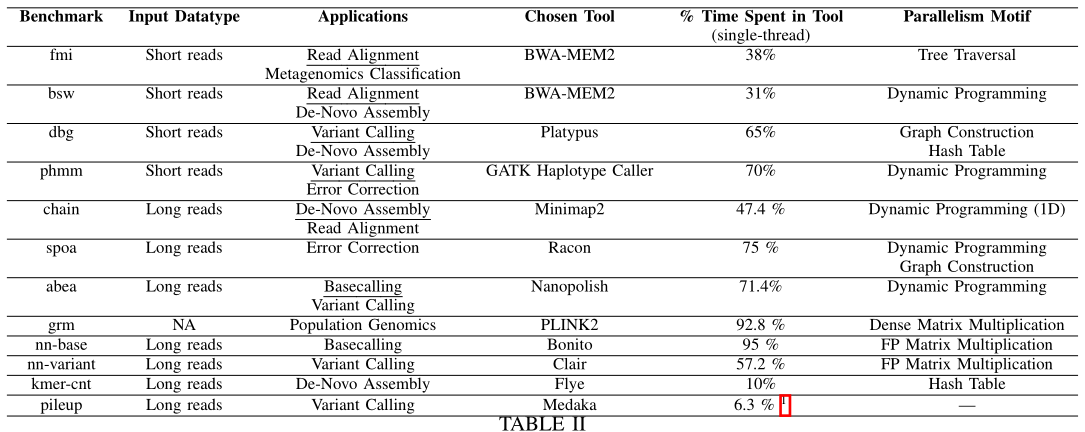
 其中CPU计算有 fmi、bsw、dbg、pairHMM、chain、poa、grm、pileup、kmer-cnt,GPU计算有nn-base、nn-variant、abea。
其中CPU计算有 fmi、bsw、dbg、pairHMM、chain、poa、grm、pileup、kmer-cnt,GPU计算有nn-base、nn-variant、abea。
这里我们主要关注CPU计算。
计算特点¶
计算分类¶
可以看到这些计算过程的并行模式都基于动态规划(dynamic programming)
数据并行¶
大部分计算过程,都可以基于 read 和 genome region 进行并行计算,由于任务大小和数据特性的多样性,使得数据并行计算的充分利用面临挑战。
指令集¶
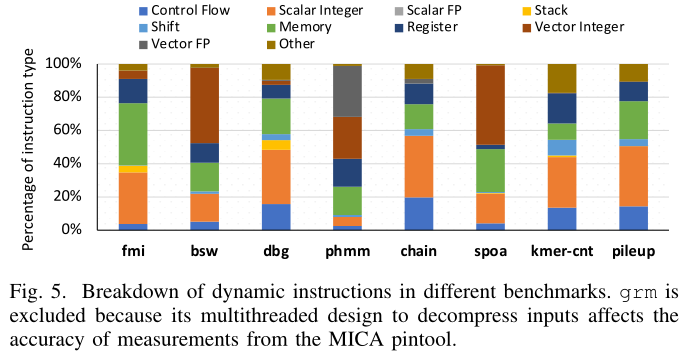
pairHMM 和 grm 计算会用到单精度浮点计算,其它计算均为整型计算;phmm、bsw和spoa受益于SIMD向量化,并且具有高比例的向量计算指令。
访存和 cache miss¶
fmi 和 kmer-cnt 对内存带宽需求较高,同时具有较高的cache miss。其它计算过程对内存带宽和缓存需求不高。


多核扩展性¶
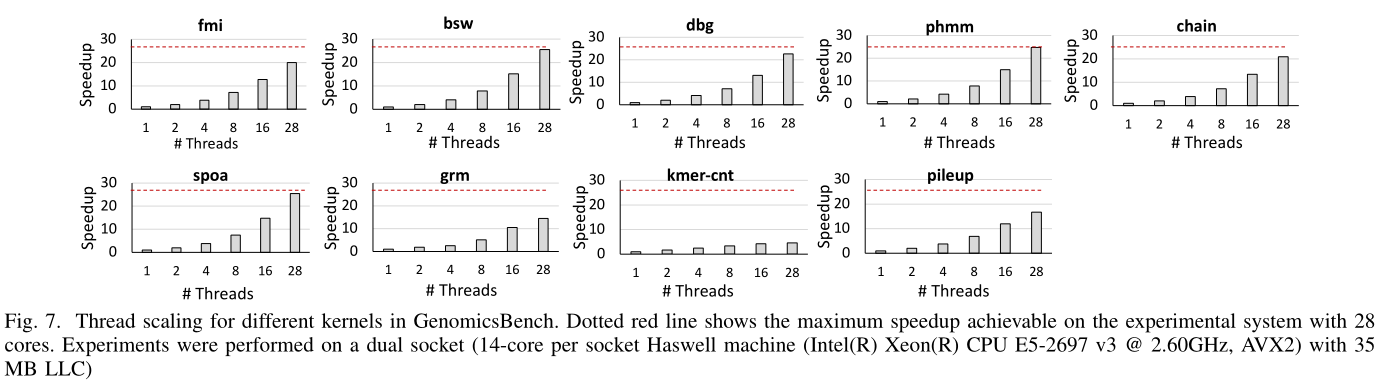
大部分计算过程具有良好的多核扩展性(bsw, dbg, phmm, spoa),fmi 和 chain 几乎线性扩展,kmer-cnt 和 pileup 受限于随机访存,其扩展性受限。
使用¶
GenomicsBench 编译容易报错,为了保持在各个系统中都能有统一的运行环境,将其封装到了docker 中,https://hub.docker.com/r/hpcbiotools/genomicsbench。
通过singularity下载:
$ singularity pull docker://hpcbiotools/genomicsbench:v1
作者提供了2个数据集:small和large,测试使用36线程满负载运行时间分别大约耗时1分钟、5分钟,数据集下载:
wget https://genomicsbench.eecs.umich.edu/input-datasets.tar.gz
运行:
# 运行large dataset,使用36线程
$ singularity exec -B input-datasets/:/opt/data/ genomicsbench_v1.sif time run-large.sh 36
# 运行small dataset,使用36线程
$ singularity exec -B input-datasets/:/opt/data/ genomicsbench_v1.sif time run-small.sh 36
结果¶
large dataset¶
| 软件 | 线程 | fmi | bsw | phmm | dbg | chain | poa | kmer-cnt | pileup | plink2 grm | total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| intel 6150 | 36(满载) | 47 | 17 | 6 | 42 | 95 | 42 | 13 | 12 | 14 | 289 |
| intel 5115 | 40(超线程,满载) | 66 | 32 | 6 | 48 | 86 | 65 | 20 | 16 | 27 | 367 |
| intel 6240 | 72 (超线程,满载) | 43 | 23 | 5 | 62 | 76 | 59 | 18 | 24 | 18 | |
| ### small dataset | |||||||||||
| 软件 | 线程 | fmi | bsw | phmm | dbg | chain | poa | kmer-cnt | pileup | plink2 grm | total |
| ------ | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| intel 6150 | 1 | 39 | 2 | 11 | 10 | 8 | 94 | 3 | 9 | 34 | 220 |
| intel 5115 | 1 | 73 | 2 | 10 | 9 | 9 | 71 | 14 | 9 | 31 | 170 |
| intel 6240 | 1 | 49 | 2 | 9 | 9 | 6 | 66 | 11 | 8 | 30 | 189 |
本站总访问量 次