单细胞教程
本文翻译自 https://github.com/quadbio/scRNAseq_analysis_vignette/blob/master/Tutorial.md
介绍¶
针对 scRNA-seq 测序数据分析开发了多种工具和分析框架,包括但不限于 Seurat(R语言开发) 和 scanpy (python开发),这两个工具提供了丰富的函数和参数集,满足大部分单细胞RNA测序数据的常规分析需求。但这些分析框架不能涵盖所有分析人员感兴趣的分析方法,因此了解其它用于单细胞RNA测序数据分析的工具也非常重要。
本教程主要面向初学者,主要还是介绍了如何在使用Seurat来分析单细胞RNA测序数据,教程最后还提到了一些具有其它分析功能的工具,如presto、destiny、Harmony、simspec等。
准备¶
本教程假设测序数据预处理步骤(包括碱基检出、比对和读数计数)已完成。10x Genomics 拥有自己的分析管道 CellRanger,用于处理使用 10x Genomics Chromium 单细胞基因表达解决方案生成的数据。在 CellRanger 流程运行完成后,会生成一个计数矩阵。如果使用的 scRNA-seq 数据是使用其他技术产生的(如 Smart-Seq2 ),则 CellRanger 可能不适用,须用另外合适的解决方案来生成计数矩阵。
作为本教程的一部分,我们提供两个数据集(DS1和DS2),均使用10x Genomics 生成并使用 Cell Ranger进行预处理。它们都是人类大脑类器官的公开scRNA-seq 数据,数据来源于文章 Organoid single-cell genomic atlas uncovers human-specific features of brain development。本教程的第一部分基于 DS1,包括大部分通用分析流程,而第二部分则基于这两个数据集,重点介绍数据集成和批量效应校正。
作为练习,可以在基于 DS2 练习通用分析流程。
数据下载地址
https://github.com/quadbio/scRNAseq_analysis_vignette/tree/master/data
10X Genomics单细胞转录组测序数据使用cellranger流程跑完后会生成3个文件 barcodes.tsv.gz,features.tsv.gz, matrix.mtx.gz。
barcodes.tsv.gz:包含了每个单细胞的条形码信息,每行代表一个细胞$ zcat barcodes.tsv.gz |head AAACCTGAGAATGTTG-1 AAACCTGAGAGCCTAG-1 AAACCTGAGTAATCCC-1 AAACCTGCACACTGCG-1 AAACCTGCATCGGAAG-1 AAACCTGGTGGTAACG-1 AAACCTGGTGTAACGG-1 AAACCTGTCCTATGTT-1 AAACGGGAGTGTACTC-1 AAACGGGAGTGTTTGC-1 # 有 4672 个细胞 $ zcat barcodes.tsv.gz |wc -l 4672features.tsv.gz:包含了全部的基因信息,每一行是一个基因,每一列代表基因对应的属性$ zcat features.tsv.gz |head ENSG00000243485 RP11-34P13.3 Gene Expression ENSG00000237613 FAM138A Gene Expression ENSG00000186092 OR4F5 Gene Expression ENSG00000238009 RP11-34P13.7 Gene Expression ENSG00000239945 RP11-34P13.8 Gene Expression ENSG00000239906 RP11-34P13.14 Gene Expression ENSG00000241599 RP11-34P13.9 Gene Expression ENSG00000279928 FO538757.3 Gene Expression ENSG00000279457 FO538757.2 Gene Expression ENSG00000228463 AP006222.2 Gene Expression # 有 33694 个基因 $ zcat features.tsv.gz |wc -l 33694matrix.mtx.gz:稀疏矩阵文件,包含了每个单细胞的基因表达信息$ zcat matrix.mtx.gz |head %%MatrixMarket matrix coordinate integer general # 包含一个标识符和四个文本域 %metadata_json: {"software_version": "cellranger-7.0.0", "format_version": 2} # 注释 33694 4672 11721086 # 矩阵大小,其中33694是基因数(行),4672是细胞数(列),11721086是矩阵中非0的数,也是接下来的行数 9 1 1 # 第一个数字对应是features.tsv.gz中,第9行的基因,第二个数字对应的是barcodes.tsv.gz中第1行的细胞,第三个数字1表示的是reads count 10 1 1 50 1 2 60 1 3 61 1 1 62 1 1 73 1 2 87 1 2 # 去掉注释说明信息,就是表达矩阵的非零的数量 $ zcat matrix.mtx.gz |wc -l 11721088
cellranger 流程跑完后,理论上应该生成一个巨大的二维表达矩阵,列为每个细胞、行为基因、值为reads count,与传统的 bulk RNA-Seq 表达矩阵类似,只不过bulk RNA-Seq 表达矩阵的列为样本。结合上面的数据,原始矩阵规模应该为 33694*4672 (行*列),矩阵元素有 157418368 个,由于大部分基因不会在所有细胞中都表达,因此这个矩阵中绝大多数的数值为0,根据cellranger跑出来的结果计算 非零数/矩阵元素 11721086/157418368=0.074,即非零数只有 7.4%,非常经典的稀疏矩阵。
如果将上面这个稀疏矩阵的所有0都表示出来,文件会非常大,计算时也会消耗大量的内存,因此稀疏矩阵就有专门的存储格式和计算算法。cellranger采用的存储格式为 dgTMatrix,是最简单的稀疏矩阵存储方式,采用三元组(row, col, data)(或称为ijv format)的形式来存储矩阵中非零元素的信息,如下所示。cellranger生成的3个文件中,matrix.mtx.gz 为 dgTMatrix 格式的表达矩阵,features.tsv.gz 和 barcodes.tsv.gz 分别为矩阵的行名和列名。
https://img-blog.csdnimg.cn/20190825163650848.gif
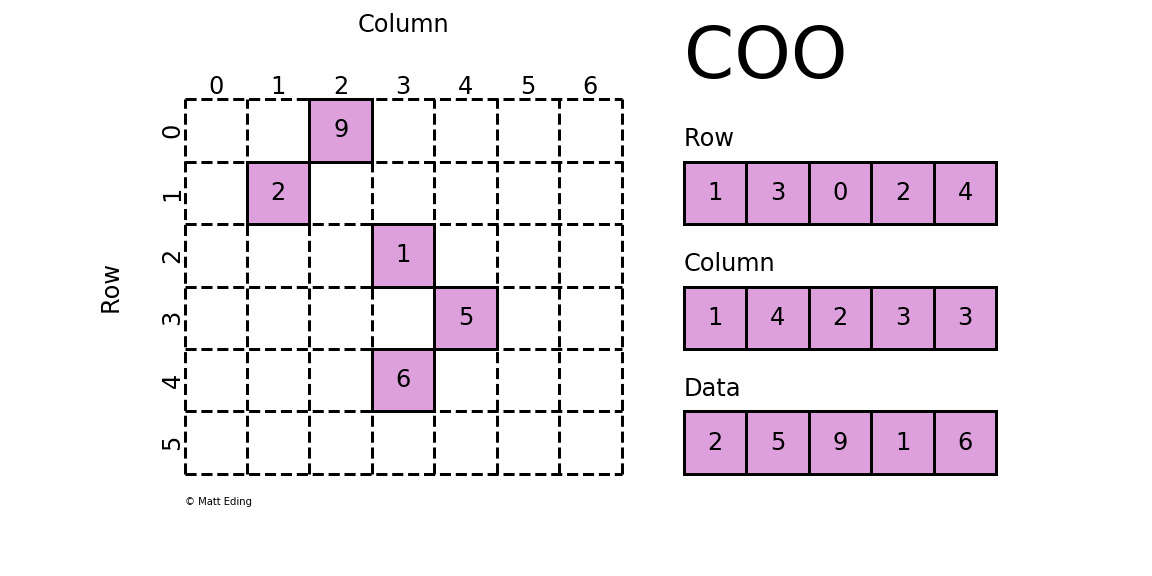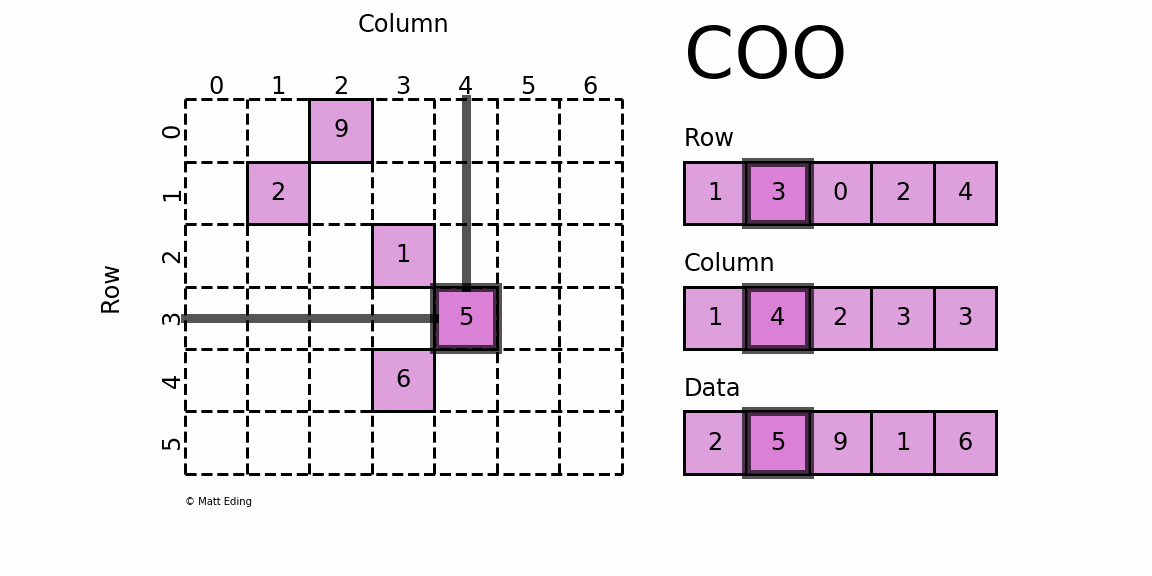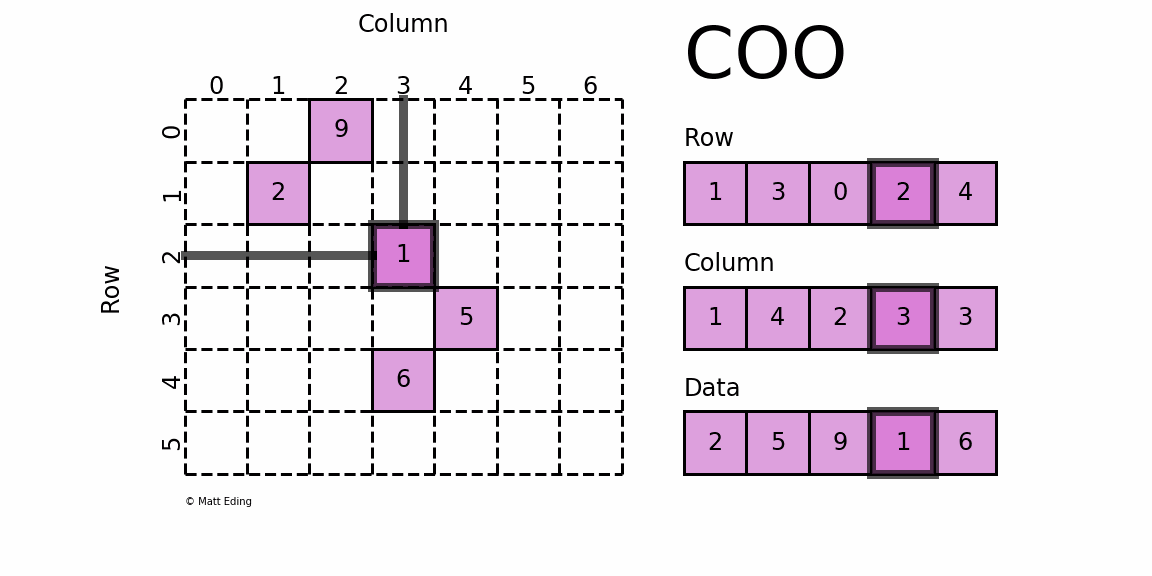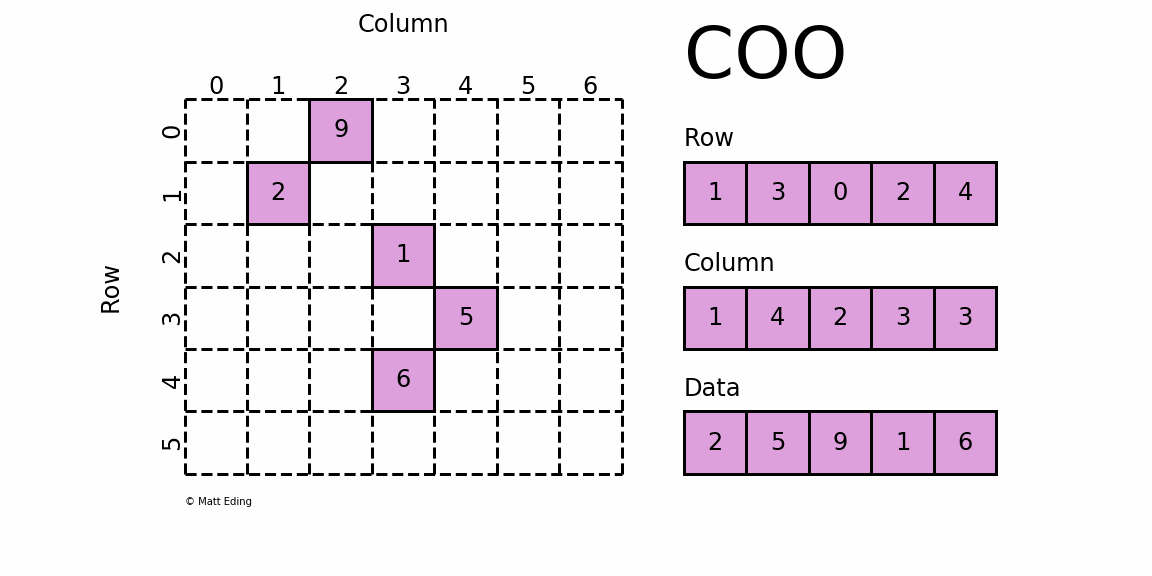{kind=link}
Seurat中单细胞稀疏数据存储采用dgCMatrix,因此用Seurat分析Cellranger输出的数据必然要先做稀疏矩阵格式的转换,而 Seurat::Read10X 函数的核心实现就是这个, Seurat::Read10X 函数会生成带有行列名的dgCMatrix。
参考:
第一部分:基本分析¶
0. 导入 Seurat 包¶
library(Seurat)
install.packages("Seurat")。 确认版本
> packageVersion("Seurat")
[1] ‘4.3.0’
1. 创建 Seurat 对象¶
Seurat 实现了一种名为Seurat的新数据类型。它允许 Seurat 存储整个分析过程中的所有步骤和结果。因此,第一步是读入数据并创建 Seurat 对象。Seurat 针对使用 10x Genomics 平台生成的数据提供了一个简单的解决方案。
counts <- Read10X(data.dir = "data/DS1/")
seurat <- CreateSeuratObject(counts, project="DS1")
Read10X 函数的作用是读取矩阵并分别用基因符号和细胞条形码重命名其行名称和列名称。如果数据不是使用 10x 平台生成时,可以手动读取数据。
library(Matrix)
counts <- readMM("data/DS1/matrix.mtx.gz")
barcodes <- read.table("data/DS1/barcodes.tsv.gz", stringsAsFactors=F)[,1]
features <- read.csv("data/DS1/features.tsv.gz", stringsAsFactors=F, sep="\t", header=F)
rownames(counts) <- make.unique(features[,2])
colnames(counts) <- barcodes
seurat <- CreateSeuratObject(counts, project="DS1")
CreateSeuratObj 函数有一些其他的选项,如 min.cells 和 min.features。使用这2个参数可以对数据进行初步过滤,从一开始就删除只有在少量细胞中表达的所有基因(min.features),以及只有少量基因表达的细胞(min.cells),这里使用默认值,即保留所有细胞和基因。
> dim(seurat)
[1] 33694 4672
seurat 对象结构¶
查看Seurat对象
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay' [package "SeuratObject"] with 8 slots
.. .. .. ..@ counts :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. ..@ factors : list()
.. .. .. ..@ data :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. ..@ factors : list()
.. .. .. ..@ scale.data : num[0 , 0 ]
.. .. .. ..@ key : chr "rna_"
.. .. .. ..@ assay.orig : NULL
.. .. .. ..@ var.features : logi(0)
.. .. .. ..@ meta.features:'data.frame': 33694 obs. of 0 variables
.. .. .. ..@ misc : list()
..@ meta.data :'data.frame': 4672 obs. of 3 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4672] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA: int [1:4672] 3287 3399 1558 2015 2322 2247 1542 2907 3472 3033 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr(*, "names")= chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs : list()
..@ neighbors : list()
..@ reductions : list()
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 4 1 3
..@ commands : list()
..@ tools : list()
>
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay5' [package "SeuratObject"] with 8 slots
.. .. .. ..@ layers :List of 1
.. .. .. .. ..$ counts:Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:11721086] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4673] 0 3287 6686 8244 10259 12581 14828 16370 19277 22749 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4672
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:11721086] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. ..@ cells :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:4672, 1] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. .. ..$ : chr "counts"
.. .. .. .. .. ..$ dim : int [1:2] 4672 1
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. ..$ : chr "counts"
.. .. .. ..@ features :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:33694, 1] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. .. ..$ : chr "counts"
.. .. .. .. .. ..$ dim : int [1:2] 33694 1
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr "counts"
.. .. .. ..@ default : int 1
.. .. .. ..@ assay.orig: chr(0)
.. .. .. ..@ meta.data :'data.frame': 33694 obs. of 0 variables
.. .. .. ..@ misc :List of 1
.. .. .. .. ..$ calcN: logi TRUE
.. .. .. ..@ key : chr "rna_"
..@ meta.data :'data.frame': 4672 obs. of 3 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4672] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA: int [1:4672] 3287 3399 1558 2015 2322 2247 1542 2907 3472 3033 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..- attr(*, "names")= chr [1:4672] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs : list()
..@ neighbors : list()
..@ reductions : list()
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 5 0 1
..@ commands : list()
..@ tools : list()
>
> str(seurat)
Formal class 'Seurat' [package "SeuratObject"] with 13 slots
..@ assays :List of 1
.. ..$ RNA:Formal class 'Assay5' [package "SeuratObject"] with 8 slots
.. .. .. ..@ layers :List of 3
.. .. .. .. ..$ counts :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:10738613] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4318] 0 3287 6686 8244 10259 12581 14123 17030 20502 23535 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4317
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:10738613] 1 1 2 3 1 1 2 2 1 1 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. .. ..$ data :Formal class 'dgCMatrix' [package "Matrix"] with 6 slots
.. .. .. .. .. .. ..@ i : int [1:10738613] 8 9 49 59 60 61 72 86 107 139 ...
.. .. .. .. .. .. ..@ p : int [1:4318] 0 3287 6686 8244 10259 12581 14123 17030 20502 23535 ...
.. .. .. .. .. .. ..@ Dim : int [1:2] 33694 4317
.. .. .. .. .. .. ..@ Dimnames:List of 2
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. .. ..$ : NULL
.. .. .. .. .. .. ..@ x : num [1:10738613] 0.468 0.468 0.786 1.026 0.468 ...
.. .. .. .. .. .. ..@ factors : list()
.. .. .. .. ..$ scale.data: num [1:3000, 1:4317] -0.259 -0.169 -0.119 -1.146 -0.306 ...
.. .. .. ..@ cells :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:4317, 1:3] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. .. .. ..$ dim : int [1:2] 4317 3
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. ..@ features :Formal class 'LogMap' [package "SeuratObject"] with 1 slot
.. .. .. .. .. ..@ .Data: logi [1:33694, 1:3] TRUE TRUE TRUE TRUE TRUE TRUE ...
.. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. .. .. ..$ dim : int [1:2] 33694 3
.. .. .. .. .. ..$ dimnames:List of 2
.. .. .. .. .. .. ..$ : chr [1:33694] "RP11-34P13.3" "FAM138A" "OR4F5" "RP11-34P13.7" ...
.. .. .. .. .. .. ..$ : chr [1:3] "counts" "data" "scale.data"
.. .. .. ..@ default : int 1
.. .. .. ..@ assay.orig: chr(0)
.. .. .. ..@ meta.data :'data.frame': 33694 obs. of 8 variables:
.. .. .. .. ..$ vf_vst_counts_mean : num [1:33694] 0.000232 0 0 0.001853 0 ...
.. .. .. .. ..$ vf_vst_counts_variance : num [1:33694] 0.000232 0 0 0.00185 0 ...
.. .. .. .. ..$ vf_vst_counts_variance.expected : num [1:33694] 0.000232 0 0 0.001939 0 ...
.. .. .. .. ..$ vf_vst_counts_variance.standardized: num [1:33694] 1 0 0 0.954 0 ...
.. .. .. .. ..$ vf_vst_counts_variable : logi [1:33694] FALSE FALSE FALSE FALSE FALSE FALSE ...
.. .. .. .. ..$ vf_vst_counts_rank : int [1:33694] NA NA NA NA NA NA NA NA NA NA ...
.. .. .. .. ..$ var.features : chr [1:33694] NA NA NA NA ...
.. .. .. .. ..$ var.features.rank : int [1:33694] NA NA NA NA NA NA NA NA NA NA ...
.. .. .. ..@ misc : Named list()
.. .. .. ..@ key : chr "rna_"
..@ meta.data :'data.frame': 4317 obs. of 6 variables:
.. ..$ orig.ident : Factor w/ 1 level "DS1": 1 1 1 1 1 1 1 1 1 1 ...
.. ..$ nCount_RNA : num [1:4317] 16752 13533 3098 5158 6966 ...
.. ..$ nFeature_RNA : int [1:4317] 3287 3399 1558 2015 2322 1542 2907 3472 3033 3471 ...
.. ..$ percent.mt : num [1:4317] 1.1 1.95 2.45 3.76 2.18 ...
.. ..$ RNA_snn_res.1 : Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
.. ..$ seurat_clusters: Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
..@ active.assay: chr "RNA"
..@ active.ident: Factor w/ 15 levels "0","1","2","3",..: 11 11 2 2 9 10 8 11 3 11 ...
.. ..- attr(*, "names")= chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
..@ graphs :List of 2
.. ..$ RNA_nn :Formal class 'Graph' [package "SeuratObject"] with 7 slots
.. .. .. ..@ assay.used: chr "RNA"
.. .. .. ..@ i : int [1:86340] 0 1 7 246 428 885 899 1167 1222 1505 ...
.. .. .. ..@ p : int [1:4318] 0 32 56 90 117 131 156 178 215 223 ...
.. .. .. ..@ Dim : int [1:2] 4317 4317
.. .. .. ..@ Dimnames :List of 2
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. ..@ x : num [1:86340] 1 1 1 1 1 1 1 1 1 1 ...
.. .. .. ..@ factors : list()
.. ..$ RNA_snn:Formal class 'Graph' [package "SeuratObject"] with 7 slots
.. .. .. ..@ assay.used: chr "RNA"
.. .. .. ..@ i : int [1:289851] 0 1 7 246 346 428 885 899 986 1167 ...
.. .. .. ..@ p : int [1:4318] 0 54 128 226 280 338 406 468 540 602 ...
.. .. .. ..@ Dim : int [1:2] 4317 4317
.. .. .. ..@ Dimnames :List of 2
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. ..@ x : num [1:289851] 1 0.25 0.379 0.333 0.111 ...
.. .. .. ..@ factors : list()
..@ neighbors : list()
..@ reductions :List of 3
.. ..$ pca :Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:50] 22.22 17.76 -9.79 -10.9 -6.09 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:50] "PC_1" "PC_2" "PC_3" "PC_4" ...
.. .. .. ..@ feature.loadings : num [1:3000, 1:50] -0.01713 0.05147 0.05231 -0.00717 0.05284 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:3000] "SST" "UBE2C" "CENPF" "TAC3" ...
.. .. .. .. .. ..$ : chr [1:50] "PC_1" "PC_2" "PC_3" "PC_4" ...
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi FALSE
.. .. .. ..@ stdev : num [1:50] 12.55 8.09 6.27 5.58 5 ...
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc :List of 1
.. .. .. .. ..$ total.variance: num 2454
.. .. .. ..@ key : chr "PC_"
.. ..$ tsne:Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:2] 33.54 32.67 -13.6 -7.95 2.93 ...
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:2] "tSNE_1" "tSNE_2"
.. .. .. ..@ feature.loadings : num[0 , 0 ]
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi TRUE
.. .. .. ..@ stdev : num(0)
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc : list()
.. .. .. ..@ key : chr "tSNE_"
.. ..$ umap:Formal class 'DimReduc' [package "SeuratObject"] with 9 slots
.. .. .. ..@ cell.embeddings : num [1:4317, 1:2] -7.686 -7.497 5.529 5.675 -0.104 ...
.. .. .. .. ..- attr(*, "scaled:center")= num [1:2] 0.257 -1.631
.. .. .. .. ..- attr(*, "dimnames")=List of 2
.. .. .. .. .. ..$ : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. .. ..$ : chr [1:2] "umap_1" "umap_2"
.. .. .. ..@ feature.loadings : num[0 , 0 ]
.. .. .. ..@ feature.loadings.projected: num[0 , 0 ]
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ global : logi TRUE
.. .. .. ..@ stdev : num(0)
.. .. .. ..@ jackstraw :Formal class 'JackStrawData' [package "SeuratObject"] with 4 slots
.. .. .. .. .. ..@ empirical.p.values : num[0 , 0 ]
.. .. .. .. .. ..@ fake.reduction.scores : num[0 , 0 ]
.. .. .. .. .. ..@ empirical.p.values.full: num[0 , 0 ]
.. .. .. .. .. ..@ overall.p.values : num[0 , 0 ]
.. .. .. ..@ misc : list()
.. .. .. ..@ key : chr "umap_"
..@ images : list()
..@ project.name: chr "DS1"
..@ misc : list()
..@ version :Classes 'package_version', 'numeric_version' hidden list of 1
.. ..$ : int [1:3] 5 0 1
..@ commands :List of 8
.. ..$ NormalizeData.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "NormalizeData.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:06:45"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "NormalizeData(seurat)"
.. .. .. ..@ params :List of 5
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ normalization.method: chr "LogNormalize"
.. .. .. .. ..$ scale.factor : num 10000
.. .. .. .. ..$ margin : num 1
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ FindVariableFeatures.RNA:Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindVariableFeatures.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:06:52"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindVariableFeatures(seurat, nfeatures = 3000)"
.. .. .. ..@ params :List of 12
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ selection.method : chr "vst"
.. .. .. .. ..$ loess.span : num 0.3
.. .. .. .. ..$ clip.max : chr "auto"
.. .. .. .. ..$ mean.function :function (mat, display_progress)
.. .. .. .. ..$ dispersion.function:function (mat, display_progress)
.. .. .. .. ..$ num.bin : num 20
.. .. .. .. ..$ binning.method : chr "equal_width"
.. .. .. .. ..$ nfeatures : num 3000
.. .. .. .. ..$ mean.cutoff : num [1:2] 0.1 8
.. .. .. .. ..$ dispersion.cutoff : num [1:2] 1 Inf
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ ScaleData.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "ScaleData.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:07:45"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "ScaleData(seurat)"
.. .. .. ..@ params :List of 9
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ model.use : chr "linear"
.. .. .. .. ..$ use.umi : logi FALSE
.. .. .. .. ..$ do.scale : logi TRUE
.. .. .. .. ..$ do.center : logi TRUE
.. .. .. .. ..$ scale.max : num 10
.. .. .. .. ..$ block.size : num 1000
.. .. .. .. ..$ min.cells.to.block: num 3000
.. .. .. .. ..$ verbose : logi TRUE
.. ..$ RunPCA.RNA :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunPCA.RNA"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:07:56"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunPCA(seurat, npcs = 50)"
.. .. .. ..@ params :List of 10
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ npcs : num 50
.. .. .. .. ..$ rev.pca : logi FALSE
.. .. .. .. ..$ weight.by.var : logi TRUE
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ ndims.print : int [1:5] 1 2 3 4 5
.. .. .. .. ..$ nfeatures.print: num 30
.. .. .. .. ..$ reduction.name : chr "pca"
.. .. .. .. ..$ reduction.key : chr "PC_"
.. .. .. .. ..$ seed.use : num 42
.. ..$ RunTSNE :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunTSNE"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:09:06"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunTSNE(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 8
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ cells : chr [1:4317] "AAACCTGAGAATGTTG-1" "AAACCTGAGAGCCTAG-1" "AAACCTGAGTAATCCC-1" "AAACCTGCACACTGCG-1" ...
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ seed.use : num 1
.. .. .. .. ..$ tsne.method : chr "Rtsne"
.. .. .. .. ..$ dim.embed : num 2
.. .. .. .. ..$ reduction.name: chr "tsne"
.. .. .. .. ..$ reduction.key : chr "tSNE_"
.. ..$ RunUMAP.RNA.pca :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "RunUMAP.RNA.pca"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:09:59"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "RunUMAP(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 25
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ slot : chr "data"
.. .. .. .. ..$ umap.method : chr "uwot"
.. .. .. .. ..$ return.model : logi FALSE
.. .. .. .. ..$ n.neighbors : int 30
.. .. .. .. ..$ n.components : int 2
.. .. .. .. ..$ metric : chr "cosine"
.. .. .. .. ..$ learning.rate : num 1
.. .. .. .. ..$ min.dist : num 0.3
.. .. .. .. ..$ spread : num 1
.. .. .. .. ..$ set.op.mix.ratio : num 1
.. .. .. .. ..$ local.connectivity : int 1
.. .. .. .. ..$ repulsion.strength : num 1
.. .. .. .. ..$ negative.sample.rate: int 5
.. .. .. .. ..$ uwot.sgd : logi FALSE
.. .. .. .. ..$ seed.use : int 42
.. .. .. .. ..$ angular.rp.forest : logi FALSE
.. .. .. .. ..$ densmap : logi FALSE
.. .. .. .. ..$ dens.lambda : num 2
.. .. .. .. ..$ dens.frac : num 0.3
.. .. .. .. ..$ dens.var.shift : num 0.1
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ reduction.name : chr "umap"
.. ..$ FindNeighbors.RNA.pca :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindNeighbors.RNA.pca"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:10:54"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindNeighbors(seurat, dims = 1:20)"
.. .. .. ..@ params :List of 16
.. .. .. .. ..$ reduction : chr "pca"
.. .. .. .. ..$ dims : int [1:20] 1 2 3 4 5 6 7 8 9 10 ...
.. .. .. .. ..$ assay : chr "RNA"
.. .. .. .. ..$ k.param : num 20
.. .. .. .. ..$ return.neighbor: logi FALSE
.. .. .. .. ..$ compute.SNN : logi TRUE
.. .. .. .. ..$ prune.SNN : num 0.0667
.. .. .. .. ..$ nn.method : chr "annoy"
.. .. .. .. ..$ n.trees : num 50
.. .. .. .. ..$ annoy.metric : chr "euclidean"
.. .. .. .. ..$ nn.eps : num 0
.. .. .. .. ..$ verbose : logi TRUE
.. .. .. .. ..$ do.plot : logi FALSE
.. .. .. .. ..$ graph.name : chr [1:2] "RNA_nn" "RNA_snn"
.. .. .. .. ..$ l2.norm : logi FALSE
.. .. .. .. ..$ cache.index : logi FALSE
.. ..$ FindClusters :Formal class 'SeuratCommand' [package "SeuratObject"] with 5 slots
.. .. .. ..@ name : chr "FindClusters"
.. .. .. ..@ time.stamp : POSIXct[1:1], format: "2024-06-05 09:10:55"
.. .. .. ..@ assay.used : chr "RNA"
.. .. .. ..@ call.string: chr "FindClusters(seurat, resolution = 1)"
.. .. .. ..@ params :List of 11
.. .. .. .. ..$ graph.name : chr "RNA_snn"
.. .. .. .. ..$ cluster.name : chr "RNA_snn_res.1"
.. .. .. .. ..$ modularity.fxn : num 1
.. .. .. .. ..$ resolution : num 1
.. .. .. .. ..$ method : chr "matrix"
.. .. .. .. ..$ algorithm : num 1
.. .. .. .. ..$ n.start : num 10
.. .. .. .. ..$ n.iter : num 10
.. .. .. .. ..$ random.seed : num 0
.. .. .. .. ..$ group.singletons: logi TRUE
.. .. .. .. ..$ verbose : logi TRUE
..@ tools : list()
>
对象结构说明¶
1 assays:存储数据集的表达矩阵及相关信息。这里只有一个RNA域,对应RNA表达数据。它包含:
- counts:原始表达计数矩阵,行为基因,列为细胞。
- data: 与counts相同,用于兼容以前的seurat版本。 数据进行了VST变换后,存储在data域,而非scale_data域。
- scale.data: 标准化后的表达矩阵。
- key:表达矩阵的标识符,此处为 "rna_"。
- assay.orig:原始表达矩阵,此处为空。
- var_features:变异基因信息。如果进行筛选,例如选定了2000个变异基因,存储在var_features域
- meta.features:包含每个特征的统计量,如平均值、方差、标准化方差等,用于过滤低变异特征。
- misc:杂项,此处为空。
2 meta_data: 存储细胞元数据,如细胞类型,count数等。meta_data域包含更丰富的信息,如percent.mito,聚类标签等
- orig.ident:表示原始数据的标识符或名称。有多个数据集时,可以用来区分不同数据集。创建seurat对象时生成。
- nCount_RNA:每个细胞的UMI计数,在RNA表达矩阵中。创建seurat对象时生成。
- nFeature_RNA:每个细胞检测到的特征数(基因数),在RNA表达矩阵中。创建seurat对象时生成。
- percent.mito:每个细胞线粒体特征的百分比,用于过滤低质量细胞。数据处理过程中定义的。
- RNA_snn_res.0.5:构建RNA_snn网络图时resolution为0.5时的聚类结果。数据处理过程中定义的。
- seurat_clusters:Seurat的FindClusters命令产生的聚类结果,resolution同为0.5。数据处理过程中定义的。
这个域汇总了样品和表达数据的基本信息,以及两种不同参数下的聚类结果。
3 active.assay:设置当前使用的表达矩阵,这里是RNA。
4 active.ident:细胞ID,对应列名。active.ident设置为聚类标签,而非原始ID
5 graphs:网络图信息,用于存储PCA,tSNE等结果。
- RNA_nn:最近邻网络图。
- RNA_snn:共享最近邻网络图。
它们包含节点(细胞)与边(相似性)的信息。
6 reductions:用于存储降维结果,如PCA,UMAP的模型对象。
cell.embeddings: 细胞在低维空间的坐标。
feature.loadings:每个维度对原始特征的贡献度。只有PCA包含。
assay.used:使用的表达矩阵,这里是RNA。
global:是否是全局分析,tSNE和UMAP为TRUE。
stdev:标准差。只有PCA包含。
key:降维 techniuqes的名称,如PC_、tSNE_ 和UMAP_。
jackstraw:置换检验的结果,评估每个维度的显著性。只有PCA包含。
misc:其他信息,如PCA的总方差。
7 images:存储绘图 Output,用于再现分析过程。
8 project.name:项目名称。
9 misc:杂项,此处为空。
10 version:seurat对象的版本信息。
11 commands:存储构建seurat对象使用的所有命令,以支持再现分析过程。
ScaleData:标准化表达矩阵。
NormalizeData:对表达矩阵进行归一化。
FindVariableFeatures:找到高变异特征。
RunPCA:进行PCA分析。
RunTSNE:进行tSNE分析。
RunUMAP:进行UMAP分析。
JackStraw:进行置换检验。
ScoreJackStraw:给置换检验结果打分。
FindNeighbors:构建邻居网络图。
FindClusters:进行聚类分析。
每个命令都包含name、time.stamp、assay.used、call.string和params等域,记录命令名称、运行时间、使用的表达矩阵、命令语句和参数等信息。
12 tools:存储分析中使用的其他软件包版本信息。
访问结构对象¶
seurat对象访问有多种访问方式。
1 @:直接使用
@可以第一层slot,如:> library(Seurat) ## Attaching SeuratObject > sobj <- seurat > counts <- sobj@assays > counts <- sobj@meta.data提取
meta.data域,获得样品元数据。2 $:直接使用
$可以访问域,如:> counts <- sobj$RNA3 结合
$和@访问第三层slot:> counts <- sobj$RNA@counts提取counts域,获得原始表达矩阵。
4 使用
[[]]可以访问域,并允许域名包含特殊字符(如.),和$作用类似。> counts <- sobj[["nCount_RNA"]] > counts <- sobj[["RNA"]]5 其它方式
# 列出所有域的名称 slotNames() # 提取高变异特征 VariableFeatures() # 函数检查类定义中指定的所有槽是否存在,并且槽中的对象是否来自所需的类或该类的子类 validObject() colnames(x = seurat) Cells(x = seurat) rownames(x = seurat) ncol(x = seurat) nrow(x = seurat) dim(seurat) Idents(object = seurat) levels(x = seurat)
参考:
2. 质量控制¶
创建Seurat对象后,下一步是对数据进行质量控制。最常见的质量控制是过滤掉下面几种细胞:
检测到含有过少基因的细胞
低质量细胞或空液滴中通常只能检测到很少的基因。
检测到含有太多基因的细胞
它们可能代表双联体或多联体(即同一液滴中的两个或多个细胞,因此共享相同的细胞条形码),这种也需要过滤掉,只分析液滴中只有一个细胞的数据。
线粒体占比高的细胞
低质量/死细胞经常表现出广泛的线粒体污染。由于大多数 scRNA-seq 实验使用 oligo-T 来捕获 mRNA,因此线粒体转录本(mitochondrial transcripts) 由于缺乏 poly-A 尾巴而应该相对代表性不足,但仍然不可避免地会捕获一些线粒体转录本。同时,有证据表明有些线粒体转录本中存在作为降解标记的稳定的 poly-A 尾巴。
Seurat 在创建 Seurat 对象时会自动计算检测到的基因数量(nFeature_RNA 是检测到的基因/特征的数量;nCount_RNA 是检测到的RNA分子数量)。计算线粒体占比可以使用函数 PercentageFeatureSet() 。
人的参考基因组GTF文件中线粒体以 MT- 开头,故用这个做正则匹配,如果是其他物种需要看具体的GTF文件,不可照搬,如小鼠是 mt-。
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT[-\\.]")
PS:不存在放之四海而皆准的过滤标准,因为这些指标的正常范围在不同实验之间可能存在巨大差异,具体取决于样本来源以及试剂和测序深度。这里的一个建议是仅过滤掉异常细胞,即某些 QC 指标明显高于或低于大多数细胞的少数细胞。为此首先需要知道这些值在数据中的分布情况。
可以通过为每个指标创建小提琴图来查看分布,图中每个点代表一个细胞。
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
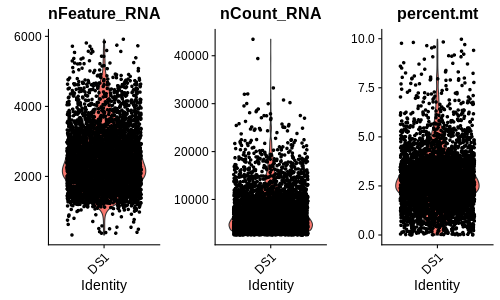
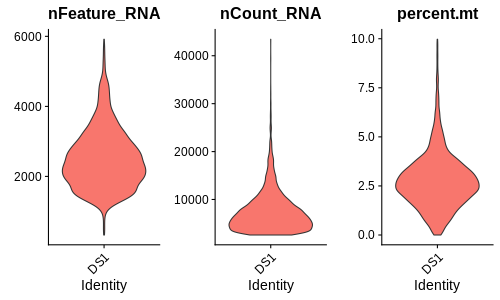
也可以画没有点的图。
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size=0)
再画一下基因数量与转录本数量、线粒体转录本数量与总的转录本数量相关性的图,可以看到前两者之间具有良好的相关性(即转录本数越多、基因数越多),后两者之间无明显相关性。
library(patchwork)
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
Note
patchwork 用于 ggplot2 生成的图的布局,Seurat 包中的绘图函数均使用 ggplot2 来生成图。
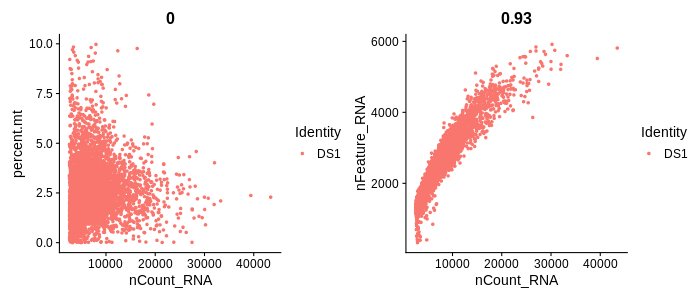
由于基因数量和转录本数量的相关性,因此只需为这些指标中的任何一个设置截止值,并结合线粒体转录本百分比的上限阈值即可进行质量控制。例如,对于这个数据集,检测到的基因数量在 500 到 5000 之间,线粒体转录物百分比低于 5% 是相当合理的,但使用不同的阈值也可以。
seurat <- subset(seurat, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 5)
一个潜在的问题是双峰的存在。
由于捕获的 RNA 数量因细胞而异,因此双联体并不总是显示出更多数量的检测到的基因或转录本。现在有几种可用的工具,旨在预测“细胞”是否确实是单峰或实际上是双峰/多重峰。例如,DoubletFinder 会首先通过对数据中的细胞进行随机平均来构建人工双峰,然后针对每个细胞测试它是否与人工双峰更相似,从而预测双峰,以判断细胞是否可能是双联体。使用举例 单细胞数据分析 | 双细胞的检测和过滤
看到一种说法,主流的单细胞转录组数据都是来源于10x技术,如果细胞数量是在5000到1万,双细胞的数量很少很少,并不会影响下游分析。
线粒体转录物百分比可能不足以过滤掉应激或不健康的细胞。
有时需要进行额外的过滤,例如 基于机器学习的预测。
核糖体
有的文章中,会过滤掉核糖体基因。建议多次反复查看线粒体核糖体基因的影响,分群前后看,不同batch看,多次质控图表里面显示它,判断它是否是一个主要因素。
单细胞测序数据中到底应该如何处理线粒体基因?这可能是个新思路
3. 标准化¶
与 bulk RNA-seq 类似,捕获的RNA量因细胞而异,因此不应直接比较细胞之间每个基因捕获的转录本的数量。因此,有必要进行标准化步骤,旨在使不同细胞之间的基因表达水平具有可比性。scRNA-seq 数据分析中最常用的标准化与 TPM(每百万次读取的转录本)的概念非常相似 - 将每个细胞的特征表达测量标准化为总表达,然后将其乘以比例因子(默认情况下为10000)。最后,对所得表达水平进行对数转换,以便表达值更好地拟合正态分布。值得一提的是,在进行对数转换之前,每个值都会添加一个伪计数,以便在细胞中检测到的零转录本的基因在对数转换后仍然呈现零值。
seurat <- NormalizeData(seurat)
在 NormalizeData() 函数中可以设置多个参数,但大多数时候默认设置就可以了。
4. 寻找差异基因(高可变基因)¶
与 bulk RNA-seq 相比,单细胞的最大优势是可以通过寻找具有不同分子特征的细胞群来研究样本的细胞异质性。然而,在尝试识别不同的细胞群时,并非每个基因都具有相同水平的信息和相同的贡献。例如,表达水平低的基因以及所有细胞中表达水平相似的基因信息量不大,可能会淡化不同细胞群之间的差异。因此,在进一步探索 scRNA-seq 数据之前,有必要进行适当的特征选择。
在 Seurat 中,或者更一般地说,在 scRNA-seq 数据分析中,此步骤通常是指识别高度可变的特征/基因,这些特征/基因是在细胞中表达水平差异最大的基因。
seurat <- FindVariableFeatures(seurat, nfeatures = 3000)
默认情况下,Seurat 计算细胞间每个基因的标准化方差,并选择前 2000 个基因作为高度可变的特征。通过提供 nfeatures 选项(此处使用前 3000 个基因),可以轻松更改高度可变特征的数量。还有一些其他的控制选项,如 mean.cutoff=c(0.0125,3),dispersion.cutoff =c(1.5,Inf)。
没有好的标准来确定要使用多少个高度可变的特征。有时需要经过一些迭代才能选择给出最清晰和可解释结果的数字。大多数情况下,2000 到 5000 之间的值就可以,使用不同的值不会对结果产生太大影响。
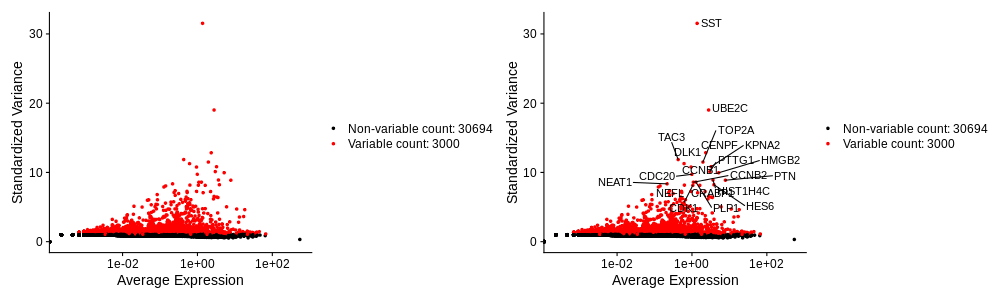
可以在变量特征图中可视化结果
top_features <- head(VariableFeatures(seurat), 20)
plot1 <- VariableFeaturePlot(seurat)
plot2 <- LabelPoints(plot = plot1, points = top_features, repel = TRUE)
plot1 + plot2
5. 数据归一化¶
由于不同基因的碱基表达水平和分布不同,如果不进行数据转换,每个基因对分析的贡献是不同的。这是我们不想要的,因为我们不希望我们的分析仅依赖于高度表达的基因。因此,使用所选特征进行归一化,归一化后的数据特征为:
改变每个基因的表达,使细胞的平均表达为0
缩放每个基因的表达,使细胞之间的方差为 1
- 这一步骤在下游分析中具有同等的权重,因此高度表达的基因不会占主导地位
结果存储在
seurat[["RNA"]]@scale.data
seurat <- ScaleData(seurat)
在此步骤中,还可以通过设置参数 var.to.regress 从数据集中删除不需要的变异源。例如,
seurat <- ScaleData(seurat, vars.to.regress = c("nFeature_RNA", "percent.mt"))
通常认为回归的变量包括检测到的基因/转录本的数量 (nFeature_RNA / nCount_RNA)、线粒体转录本百分比 (percent.mt) 和细胞周期相关变量。它试图做的是首先拟合一个线性回归模型,使用基因的标准化表达水平作为因变量,并将要回归的变量作为自变量。然后将线性模型的残差视为去除了所考虑变量的线性效应的信号。应该注意到,这个回归变量的过程极大地减慢了整个过程,并且不清楚结果是否令人满意,因为不需要的变化可能远非线性。因此,一个常见的建议是在数据探索的第一次迭代中不要执行任何回归,而是首先检查结果,如果任何不需要的变异源主导了细胞异质性,则尝试回归相应的变量并查看是否结果改善。
可选替换步骤3-5:使用 SCTransform¶
经典的 对数归一化(log-normalization) 的一个问题是将零膨胀(introduces the zero-inflation artifact)引入到 scRNA-seq 数据中。为了更好地解决这个问题,Hafemeister 和 Satija 引入了 R 包 sctransform ,它使用正则化负二项式回归模型来标准化 scRNA-seq 数据。Seurat 有一个包装函数 SCTransform 。
零膨胀现象(zero-inflated):某事件发生次数的资料中含有大量的零,即许多观察个体在单位时间、单位体积内未观察到相应事件的发生。这些资料零观测值出现的概率远远超出相同条件下标准计数模型( 如Poisson 回归和负二项回归模型) 能够预期的范围,使模型的方差远大于期望,这种现象称为零膨胀(zero-inflated)现象。在大多数细胞中,许多基因没有检测到分子,导致单细胞计数矩阵中有大量的零。
seurat <- SCTransform(seurat, variable.features.n = 3000)
variable.features.n 控制要识别的高度可变特征的数量。我们还可以添加有关要回归哪些不需要的变异来源的信息。例如, seurat <- SCTransform(seurat,
vars.to.regress = c("nFeature_RNA", "percent.mt"),
variable.features.n = 3000)
此操作结合了标准化、归一化和高可变特征选择,因此它基本上取代了上面的步骤 3-5。
使用 SCTransform 的优点包括:
聚类结果受测序深度(nCount_RNA)影响较小;
较高的细胞类型分辨率,鉴定到的细胞类型多。
缺点包括:
运行速度相对较慢。
它使得标准化表达量测数据依赖于数据本身。在标准程序中,归一化仅依赖于细胞本身;然而,在
SCTransform中,标准化过程中涉及来自同一数据集中其他细胞的信息。当需要比较多个数据集时,这可能会带来问题,因为使用SCTransform单独标准化的两个数据集的标准化表达测量结果不具有可比性。SCTransform中有一些步骤涉及随机采样以加快计算速度。这意味着SCTransform存在一定的随机性,即使应用于相同的数据集,结果每次也略有不同。
因此,应该明智地使用它。在某些情况下,建议尝试 SCTransform。例如,您可能在两种条件下的细胞之间进行差异表达分析,结果显示 DE 存在显着偏差,即大量基因表达增加,但很少基因表达减少。如果两种条件的数据显示出非常不同的覆盖范围(例如,表达大量减少的条件显示检测到的基因/转录本数量显着减少),则这种有偏差的 DE 结果可能确实是标准标准化程序引入的人为因素。在这种情况下,使用 SCTransform 可能有助于减少伪影。
参考:
6. PCA 线性降维¶
原则上,在识别出高可变基因并对数据归一化后,我们就可以开始研究细胞异质性。然而强烈建议在进行任何进一步分析之前应用线性降维。进行这种降维的好处包括但不限于:
数据变得更加紧凑,因此计算速度变得更快。
由于 scRNA-seq 数据本质上是稀疏的,因此总结相关特征的测量结果可以大大增强信号的鲁棒性。
对于 scRNA-seq 数据,线性降维主要是指主成分分析(PCA)。
主成分分析(Principal component analysis,PCA),是考察多个变量间相关性的一种多元统计方法,研究如何通过少数几个主成分来揭示多个变量间的内部结构,即从原始变量中导出少数几个主成分,使它们尽可能多地保留原始变量的信息,将原来n个指标作线性或非线性组合,作为新的综合指标。
seurat <- RunPCA(seurat, npcs = 50)
npcs 参数来更改这一点。 即使如此,也不一定会使用所有计算出的 PC。确定使用多少个 PC 是一门艺术。没有什么黄金标准,每个人都有自己的理解。如果 PC 太多,变化最小的 PC 可能代表数据中的噪声并稀释信号。如果 PC 太少,可能无法捕获数据中的基本信息。最佳数量的 PC 应保留数据中的基本信息,同时尽可能多地滤除噪音。
使用肘部图方法可以帮助做出决定,它包括将解释的变化绘制为每个 PC 的函数,并选择曲线的肘部作为要使用的 PC 的数量。
ElbowPlot(seurat, ndims = ncol(Embeddings(seurat, "pca")))
Embeddings是 Seurat 中的函数,用于在给定感兴趣的降维名称的情况下获取降维结果。默认情况下,RunPCA函数将 PCA 结果存储在名为 "pca" 的嵌入中,每一列都是一个 PC。所以这里它告诉 Seurat 构建肘部图来显示计算出的所有 PC 的标准化变化
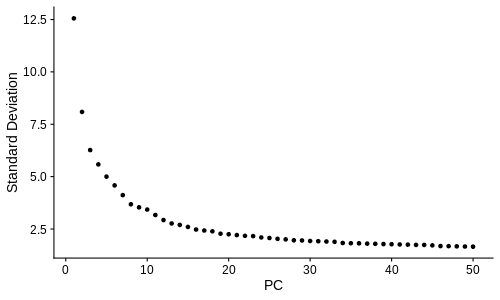
图中 y 轴显示 PC 的标准差,x 轴显示 PC 的数量,排名较高的 PC 比排名较低的 PC 解释更多的数据变化(具有更高的标准差)。然而,标准差的降低不是线性的。对于前几个 PC,肘部图的曲线急剧下降,然后减慢并变得相当平坦。人们会假设曲线的第一阶段代表与细胞群之间的生物学差异相关的"真实"信号,而第二阶段主要代表技术变化或单个细胞的随机性质。从这个角度来看,选择排名前 15 的 PC 可能是好的,而排名低于 20 的 PC 看起来完全没有必要。然而,尽管这是一个很好的参考,但它还远非完美:
精确定义曲线的肘点或转折点非常困难,因为它通常不是完美的肘部。
排名较高的PCs确实比排名较低的PCs解释了更多的变化,但更多解释的变化并不一定意味着更高的信息内容。有时,噪声中隐藏着一些有趣但微弱的信号,因此成为排名较低的 PC 的一部分。
Seurat 中实现的另一个过程称为 JackStraw ,它也可以帮助确定需要考虑多少个 PC 进行以下分析。然而,根据我们的经验,这个过程非常慢,因为它依赖于数据排列,并且它基本上不提供比肘图更多的信息。它的作用是估计每个 PC 的统计显著性,但同样,显著性 PC 并不意味着它具有丰富的信息。当细胞数量增加时,越来越多的 PC 变得具有统计上的显著性,尽管它们所解释的变异并不显着。对这种方法感兴趣的人可以看看Seurat小插图。
除了做出公正的决定外,我们还可以检查哪些基因对每一个 top PC 的贡献最大。如果人们了解所分析样本的基因和生物学特性,这可能会提供丰富的信息。它提供了了解每一个 top PC 生物学意义的机会,以方便选择那些代表有用信息的 PC。
PCHeatmap(seurat, dims = 1:20, cells = 500, balanced = TRUE, ncol = 4)

PS:不建议仅选择由“有趣”基因代表的 PC。这样做很有可能会错过有趣但意想不到的现象。
在本例中,我们将使用排名前 20 的 PC 进行以下分析。使用更多或更少的 PC 绝对没问题,在实践中有时需要反复尝试才能做出最终决定。对于大多数数据来说,PC 数在10到50之间是合理的,很多情况下不会对结论产生太大影响(但有时会,所以还是要谨慎)。
7. 非线性降维(可视化)¶
Note
非线性降维主要是为了可视化,超过3个维度无法画图表示。
线性降维既有优点也有缺点。好的一面是,每个 PC 都是基因表达的线性组合,因此 PC 的解释很简单。而且数据被压缩但没有被扭曲,因此数据中的信息大部分被保留。另一方面,坏的一面是通常需要 10 个以上的 PC 才能覆盖大部分信息。这对于大多数分析来说都很好,但对于可视化来说就不行了,因为普通人不可能跨越三个维度。
为了克服这个问题,引入了非线性降维。scRNA-seq 数据分析中最常用的非线性降维方法是t-SNE 和UMAP。两种方法都试图将每个样本放置在低维空间(2D/3D)中,使得原始空间中不同样本(这里是细胞)之间的距离或邻域关系很大程度上保留在低维空间中。这两种方法的详细数学描述见 tSNE 的视频和 UMAP 的 博客。还有更多方法可以创建其他低维嵌入以进行可视化,包括但不限于 SPRING、PHATE。
PCA 分析中的 top PC 用作 tSNE 和 UMAP 的输入数据。
seurat <- RunTSNE(seurat, dims = 1:20)
seurat <- RunUMAP(seurat, dims = 1:20)
结果可以可视化:
plot1 <- TSNEPlot(seurat)
plot2 <- UMAPPlot(seurat)
plot1 + plot2
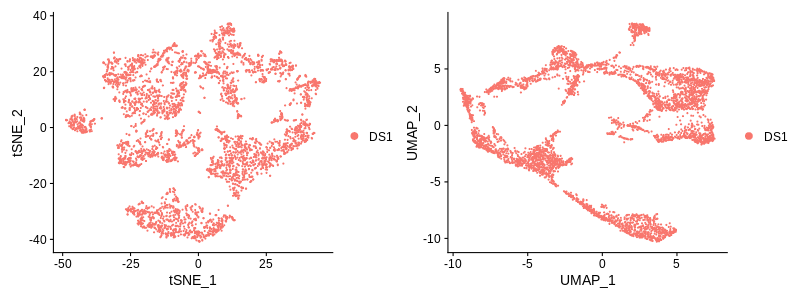
tSNE 或 UMAP 是否优于另一个是一个热门话题(如,Nikolay Oskolkov的博客 和 Kobak 和 Linderman 的文章。根据经验,它们都有各自的优点和缺点,并且都不总是比另一种效果更好。当细胞形成不同的细胞组时,tSNE 提供了很好的可视化,而当数据包含“连续体”时,UMAP 可以更好地保留轨迹状结构,例如发育和分化过程中细胞状态的连续变化。因此,最好同时尝试这两种方法,根据数据选择更合适的一种。
运行完 tSNE 或 UMAP 后,可以绘制感兴趣细胞类型的已知标记的特征图,以此来开始检查数据中是否存在某些细胞类型或细胞状态。
plot1 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "tsne")
plot2 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "umap")
plot1 / plot2
导入 patchwork 后, plot1 / plot2 生成plot1位于plot2上方的绘图布局。
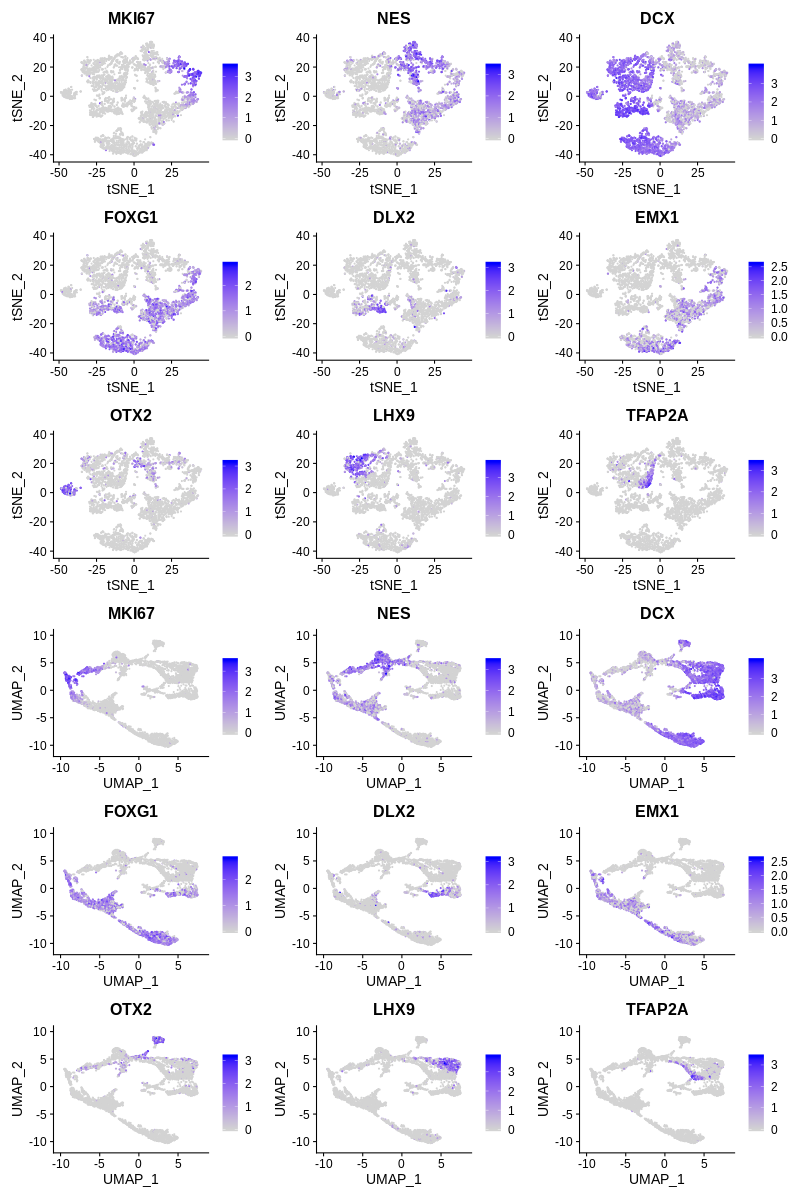
基因说明:
MKI67: a marker of G2M phase of cell cycle
NES: a neural progenitor marker
DCX: a pan-(immature) neuron marker
FOXG1: a telencephalon marker
DLX2: a ventral telencephalon marker
EMX1: a dorsal telencephalon (cortex) marker
OTX2: a midbrain and diencephalon neuron marker
LHX9: a diencephalon and midbrain neuron marker
TFAP2A: a midbrain-hindbrain boundary marker
8. 细胞聚类¶
在探索 scRNA-seq 数据时,绘制 markers 的 Feature plot 通常是一个很好的起点。然而,为了更全面地了解数据中潜在的异质性,有必要以一种无偏的方式识别细胞群体,这就是聚类的作用。原则上,任何聚类方法都可以应用于 scRNA-seq 数据,包括广泛用于 bulk RNA-seq 数据分析的方法,例如层次聚类和 k-means。然而,在实践中,这是非常困难的,因为 scRNA-seq 数据中的样本量太大(一个 10x 实验通常会给出数千个细胞,相当于 bulk RNA-Seq 的几千个样本),使用这些方法会非常慢。此外由于 scRNA-seq 数据的稀疏性,即使像 PCA 一样通过降维对数据进行去噪,不同细胞之间的差异也不像 bulk RNA-seq 数据那样被准确地量化。因此,scRNA-seq 数据分析中更常用的聚类方法是基于图的社区识别算法(graph-based community identification algorithm)。这里图是数学概念,表示一组对象,其中某些对象之间存在关联。
聚类方法基于构建细胞之间的相似性网络(图),通过计算细胞之间的相似度来识别具有相似表达模式的细胞群体。这些算法能够更好地处理单细胞RNA测序数据的固有特点,例如高维度、稀疏性和噪音。通过聚类分析,可以发现数据中的细胞亚群,并深入研究这些亚群之间的差异和生物学含义。
首先,生成细胞的k近邻网络,每个细胞首先根据其相应的 PC 值连接到距离最短的细胞,只有彼此相邻的细胞对才被视为已连接。然后计算每个细胞对之间共享邻居的比例,并用于描述两个细胞之间的连接强度,弱连接被修剪,这样就得到了共享最近邻( Shared Nearest Neighbor,SNN)网络。这在Seurat中非常简单,使用 FindNeighbors() 函数即可完成计算。
seurat <- FindNeighbors(seurat, dims = 1:20)
网络构建完成后,使用louvain社区识别算法对网络进行分析,以寻找网络中的社区,即同一组内的细胞倾向于相互连接,而不同组细胞之间的连接很稀疏。
seurat <- FindClusters(seurat, resolution = 1)
resolution 参数用于控制返回的细胞群体的大小,可以选择返回主要和粗略的细胞群体(例如,主要的细胞类型),或者更小但更精细的细胞群体(例如,细胞亚型)。常用的分辨率范围在 0.1 到 1 之间,选择哪个选项取决于分析的目的,使用高分辨率参数可以获得更精细的聚类结果,可以使用不同的分辨率参数多次运行 FindClusters() 函数以获得想要的效果。通过 Idents(seurat) 或 seurat@active.ident 可以获得最新的聚类结果,其他聚类结果也被存储为 meta.data 槽中的不同列 (seurat@meta.data )中,以便进一步的分析和解释。 接下来是使用之前生成的 tSNE 和 UMAP 数据来可视化聚类结果。
plot1 <- DimPlot(seurat, reduction = "tsne", label = TRUE)
plot2 <- DimPlot(seurat, reduction = "umap", label = TRUE)
plot1 + plot2
PS: 如果不显示簇标签,请设置 label = FALSE 或删除它(默认情况下 label 设置为 FALSE)。 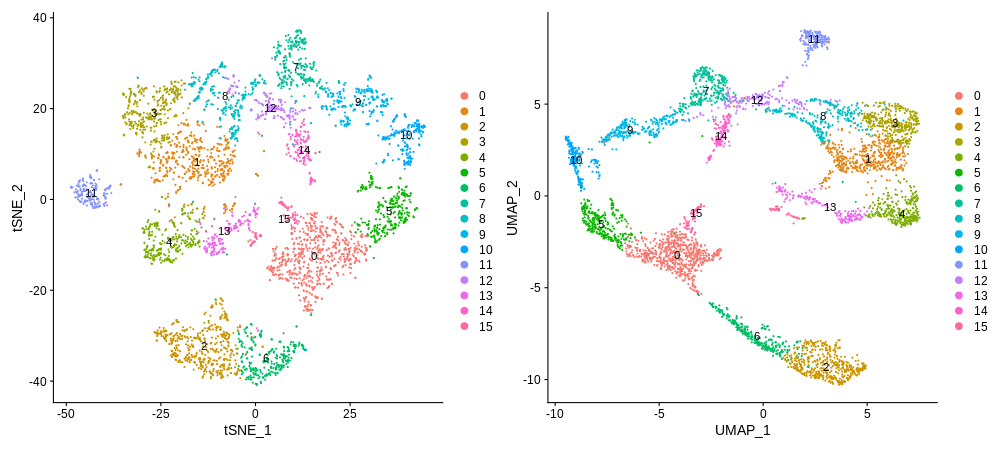
9. 细胞簇注释¶
细胞聚类为每个细胞提供了身份标签,我们可以假设具有相同标签的细胞彼此相似,因此可以被视为具有相同的细胞类型或细胞状态。下一个问题是这些细胞簇代表哪些确切的细胞类型或细胞状态。这不是一个容易回答的问题,通常没有完美的答案。有多种选择可以尝试解决此问题。例如,
检查这些聚类中的典型细胞类型和细胞状态标志物的表达;
识别每个已识别细胞簇的特征基因或标记基因。根据识别出的聚类标记基因,可以进行文献检索、富集分析或做实验(或请教周围的人)进行注释;
对于每个簇,将其基因表达谱与现有参考数据进行比较。
显然,第一种方法需要对被测量的系统有一些先验知识:需要有一份被领域内广泛接受的令人信服的markers清单。特别是对于示例数据集的系统(大脑类器官),上面列出了一些markers。更长的列表包括:
- NES / SOX2: NPC marker
- DCX: neuron marker
- FOXG1: telencephalon marker
- EMX1: dorsal telencephalon (cortical) marker
- GLI3: cortical NPC marker
- EOMES: cortical intermediate progenitor (IP) marker
- NEUROD6 / SLC17A7: dorsal excitatory (cortical) neuron marker
- BCL11B: deeper layer cortical neuron marker
- SATB2: upper layer cortical neuron marker
- RELN: Cajal-Retzius cell marker
- DLX2 / DLX5: ganglionic eminence (GE) marker
- ISL1: lateral ganglionic eminence (LGE) inhibitory neuron marker
- NKX2-1: medial ganglionic eminence (MGE) inhibitory neuron marker
- NR2F2: caudal ganglionic eminence (CGE) inhibitory neuron marker
- RSPO3 / TCF7L2 / LHX9: diencephalon marker (for different neuron subtypes)
- OTX2: midbrain marker
- HOXB2 / HOXB5: hindbrain marker
- SLC17A6: non-telencephalic excitatory neuron marker
- SLC32A1 / GAD1 / GAD2: inhibitory neuron marker
- TTR: choroid plexus marker
- GFAP: astrocyte marker
- OLIG1: oligodendrocyte precursor cell marker
- AIF1: microglia marker
- CLDN5: endothelial cell marker
- MKI67: cell cycle G2M phase marker (proliferative cells)
PS:其中许多基因不仅在列出的细胞类型中表达。例如,许多腹侧远端。神经元还表达 BCL11B,这是著名的深层皮层神经元标记。因此,在进行注释时,需要考虑markers的组合。例如,只有当 BCL11B+ 细胞也表达 EMX1 和 NEUROD6 时,它们才会被注释为更深层的皮质神经元。
最容易将感兴趣的标记基因在细胞群集中可视化的方式可能是通过热图。
ct_markers <- c("MKI67","NES","DCX","FOXG1", # G2M, NPC, neuron, telencephalon
"DLX2","DLX5","ISL1","SIX3","NKX2.1","SOX6","NR2F2", # ventral telencephalon related
"EMX1","PAX6","GLI3","EOMES","NEUROD6", # dorsal telencephalon related
"RSPO3","OTX2","LHX9","TFAP2A","RELN","HOXB2","HOXB5") # non-telencephalon related
DoHeatmap(seurat, features = ct_markers) + NoLegend()
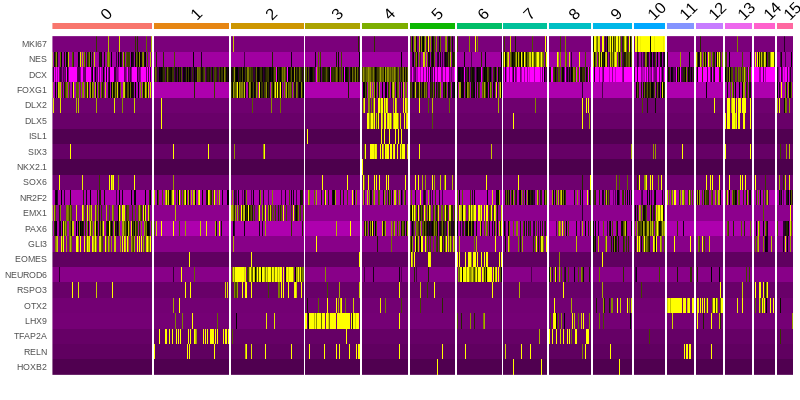
接下来,为了以更加客观的方式进行注释,首先应该确定每个已识别细胞簇的簇标记基因。在Seurat中,可以使用 FindAllMarkers() 函数来实现。该函数的作用是对每个细胞簇进行差异表达分析(使用Wilcoxon秩和检验),比较簇内的细胞与其他簇中的细胞之间的差异。
cl_markers <- FindAllMarkers(seurat, only.pos = TRUE, min.pct = 0.25, logfc.threshold = log(1.2))
library(dplyr)
cl_markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)

由于 scRNA-seq 数据样本量较大(一个细胞就是一个样本),强烈建议不仅查看 p 值,还要查看簇中基因的检出率(pct)。这就是为什么函数中有 min.pct 和 logfc.threshold 参数的原因,它们可以要求对效应大小设置阈值。
min.pct用于指定在进行差异表达基因分析时,每个簇中要包含的细胞的最小百分比。这个参数可以确保只有那些在簇中高度表示的基因被考虑,从而过滤掉在大多数细胞中表达水平较低的基因。logfc.threshold用于设置对差异表达基因的效应大小的阈值。具体来说,它定义了基因在不同簇之间的平均对数折叠变化值必须达到的最低阈值。这个阈值可以确保只有那些在群集之间具有足够大的差异表达的基因被识别出来。
通过设置这两个参数,可以更严格地控制差异表达基因分析的结果,确保只有那些具有显著差异表达且效应大小足够大的基因被识别出来。这有助于提高分析的可靠性和准确性,从而更好地理解不同簇之间的基因表达差异。
这个过程也可以使用运行速度更快的包 presto。
library(presto)
cl_markers_presto <- wilcoxauc(seurat)
cl_markers_presto %>%
filter(logFC > log(1.2) & pct_in > 20 & padj < 0.05) %>%
group_by(group) %>%
arrange(desc(logFC), .by_group=T) %>%
top_n(n = 2, wt = logFC) %>%
print(n = 40, width = Inf)
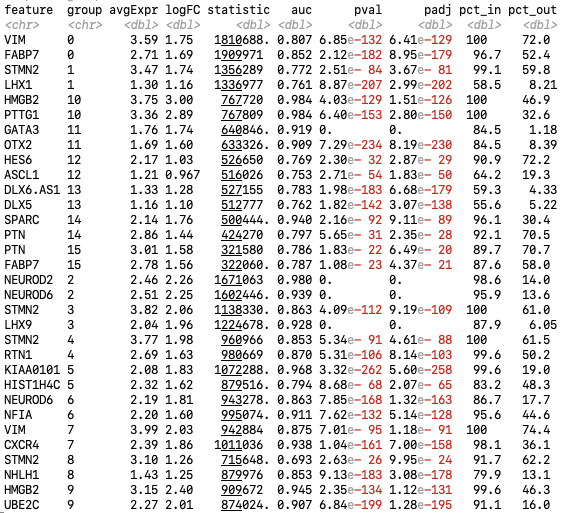
PS:最新的 presto 需要安装 DESeq2。如果 Wilcoxon 测试就足够了,使用旧版本的 presto 即可。按 devtools::install_github("immunogenomics/presto", ref = "4b96fc8") 执行。
presto 输出与 Seurat 的输出非常相似,但有一些额外的指标。
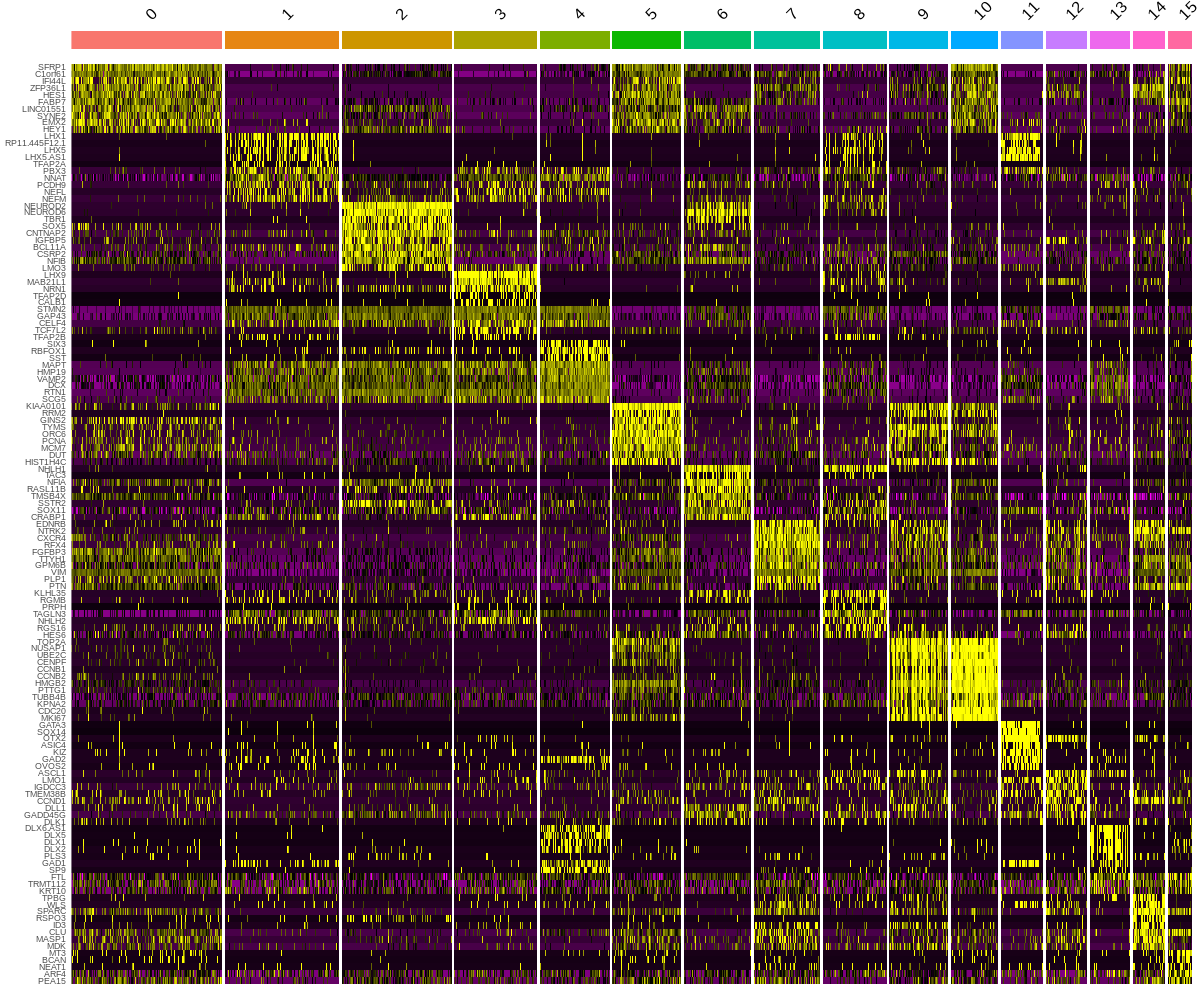
无论使用哪种方法,识别出的顶级聚类标记都可以通过热图进行可视化。
top10_cl_markers <- cl_markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(seurat, features = top10_cl_markers$gene) + NoLegend()
通过特征图或小提琴图,可以更详细地检查这些不同簇的标记。例如,可以使用 NEUROD2 和 NEUROD6 作为 cluster 2 的 strong marker。
plot1 <- FeaturePlot(seurat, c("NEUROD2","NEUROD6"), ncol = 1)
plot2 <- VlnPlot(seurat, features = c("NEUROD2","NEUROD6"), pt.size = 0)
plot1 + plot2 + plot_layout(widths = c(1, 2))
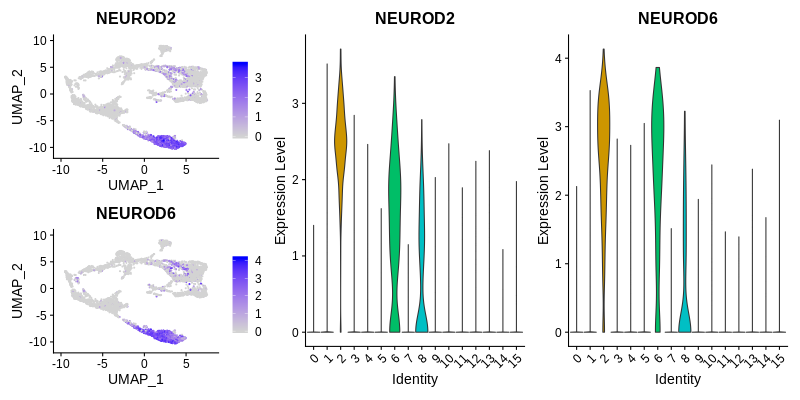
从图中来看,NEUROD2 和 NEUROD6 不仅在 cluster 2 中高度表达,而且在 cluster 6 中也高度表达。在 tSNE/UMAP 图中,这两个cluster也彼此相邻,表明它们可能代表彼此相关的细胞类型,并且都表现出强烈的背侧端脑特征。他们的分离可能代表了他们的成熟状态。cluster 6 中的神经元可能不太成熟,因为它们与 cluster 0 连接,cluster 0 可能是背侧端脑 NPCs。此外,cluster 6 显示 EOMES 的高表达。将它们放在一起,可以确信,cluster 0 6 2 都代表背侧端脑细胞,cluster 0 是祖细胞,cluster 6 是中间祖细胞,cluster 2 是神经元。
观察 cluster 0 的另一侧,连接的是 cluster 5,然后是 cluster 10。再次查看热图,发现虽然它们具有不同的标记和表达模式,但它们都呈现相似的特征背侧端脑 NPCs。最重要的是,cluster 5 和 cluster 10 中的细胞显示出细胞周期 G2M 期标记物的高表达。这表明 cluster 5 和 cluster 10 也是背侧端脑 NPC,它们与 cluster 0 的分离可能是由于它们在细胞周期阶段的差异。
有趣的是,cluster 10 5 0 6 2 中的所有这些单元在 UMAP 中形成轨迹状结构。它可能反映了神经元分化和神成熟的过程。
这就是细胞簇注释的一般方式,可能会显得太主观,太依赖个人判断。在这种情况下,还有更客观、公正的方法来进行自动化或半自动化注释。这类工具在工具不断涌现,例如 Cole Trapnell 实验室开发的 Garnett,以及 Samantha Morris 实验室开发的 Capybara。这些工具使用类似的策略,首先标准化现有 scRNA-seq 数据的细胞类型注释,使用注释数据训练一个或多个预测模型,然后将模型应用到新的数据集进行自动注释,目前这些工具都有局限性,它们的应用通常仅限于被普遍研究器官的主要细胞类型,并且它们的表现很大程度上取决于训练数据集的数据和注释质量。
值得一提的是,人们并不总是需要使用在其他 scRNA-seq 数据上训练的复杂机器学习模型来辅助细胞簇的注释。计算 scRNA-seq 数据中细胞或细胞簇的基因表达谱与大量参考数据的相关性也可以提供非常丰富的信息。一个例子是 VoxHunt,它将细胞或细胞簇的表达谱与 Allen Brain Atlas 中发育中小鼠大脑的原位杂交图谱相关联。这对于注释大脑类器官样本的 scRNA-seq 数据非常有帮助。
PS:为此,需要首先安装 voxhunt 软件包。请按照页面上的说明进行操作,并且不要忘记下载 ABA ISH 数据,页面上也有一个链接。将下面的 ABA_data 替换为下载数据的文件夹路径。
library(voxhunt)
load_aba_data('ABA_data')
genes_use <- variable_genes('E13', 300)$gene
vox_map <- voxel_map(seurat, genes_use=genes_use)
plot_map(vox_map)
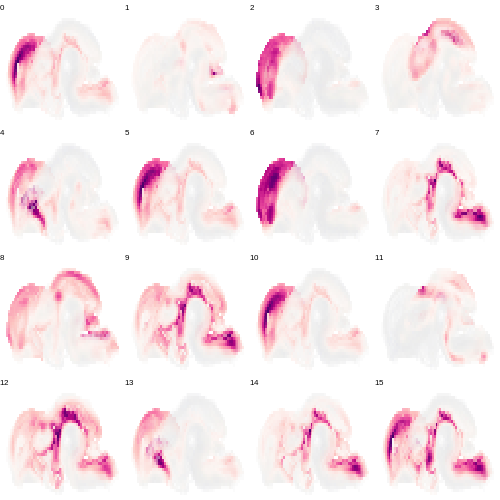
从图中我们也可以得出类似的结论,cluster 10、5、0、6、2 属于背侧端脑。最后,可以对所有 cluster 进行粗略的注释。
| Cluster | Annotation |
|---|---|
| 0 | Dorsal telen. NPC |
| 1 | Midbrain-hindbrain boundary neuron |
| 2 | Dorsal telen. neuron |
| 3 | Dien. and midbrain excitatory neuron |
| 4 | MGE-like neuron |
| 5 | G2M dorsal telen. NPC |
| 6 | Dorsal telen. IP |
| 7 | Dien. and midbrain NPC |
| 8 | Dien. and midbrain IP and excitatory early neuron |
| 9 | G2M Dien. and midbrain NPC |
| 10 | G2M dorsal telen. NPC |
| 11 | Dien. and midbrain inhibitory neuron |
| 12 | Dien. and midbrain IP and early inhibitory neuron |
| 13 | Ventral telen. neuron |
| 14 | Unknown 1 |
| 15 | Unknown 2 |
为方便展示,可以用注释替换细胞簇标签
new_ident <- setNames(c("Dorsal telen. NPC",
"Midbrain-hindbrain boundary neuron",
"Dorsal telen. neuron",
"Dien. and midbrain excitatory neuron",
"MGE-like neuron","G2M dorsal telen. NPC",
"Dorsal telen. IP","Dien. and midbrain NPC",
"Dien. and midbrain IP and excitatory early neuron",
"G2M Dien. and midbrain NPC",
"G2M dorsal telen. NPC",
"Dien. and midbrain inhibitory neuron",
"Dien. and midbrain IP and early inhibitory neuron",
"Ventral telen. neuron",
"Unknown 1",
"Unknown 2"),
levels(seurat))
seurat <- RenameIdents(seurat, new_ident)
DimPlot(seurat, reduction = "umap", label = TRUE) + NoLegend()

10. 拟时序分析¶
如上所述,我们在 UMAP 中看到的背侧端脑簇中的细胞形成的轨迹状结构可能代表了背侧端脑兴奋性神经元的分化和成熟。这可能是一个连续的过程,因此将其视为连续的轨迹而不是不同的簇更合适。在这种情况下,对这些细胞执行所谓的伪时间细胞排序或伪时间分析会提供更多信息。
迄今为止,伪时间分析的方法有相当多。常用的方法有扩散图谱(在R中的 destiny 包中实现)和 monocle。这里,我们将展示使用 destiny 对数据中的背侧端脑细胞进行伪时间分析的示例。
首先,提取感兴趣的细胞,然后重新识别子集细胞的高可变基因,因为代表背侧端脑细胞和其他细胞之间差异的基因不再提供信息。
seurat_dorsal <- subset(seurat, subset = RNA_snn_res.1 %in% c(0,2,5,6,10))
seurat_dorsal <- FindVariableFeatures(seurat_dorsal, nfeatures = 2000)
PS:如果您确定要用于子集细胞的聚类结果是最新的,则使用 seurat_dorsal <- subset(seurat, idents = c(0,2,5,6,10)) 会得到相同的结果。通过这种方式,它会在 Seurat 对象中查找聚类结果中的细胞,该结果存储为 seurat@active.ident 。在元数据表( seurat@meta.data )中,还有一个名为 seurat_clusters 的列,它显示最新的聚类结果,并且每次完成新的聚类时都会更新。 您可能已经注意到,有两个背侧端脑 NPC 簇与第三个簇分开,因为它们处于细胞周期的不同阶段。由于我们对分化和成熟过程中的一般分子变化感兴趣,因此细胞周期的变化可能会严重干扰分析。我们可以尝试通过从已识别的高可变基因列表中排除细胞周期相关基因来减少细胞周期效应。
VariableFeatures(seurat) <- setdiff(VariableFeatures(seurat), unlist(cc.genes))
cc.genes 是Seurat在导入包时自动导入的列表。它包括这篇文章中报道的基因。 然后我们可以通过创建新的 UMAP 并绘制一些特征图来检查数据的外观。
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
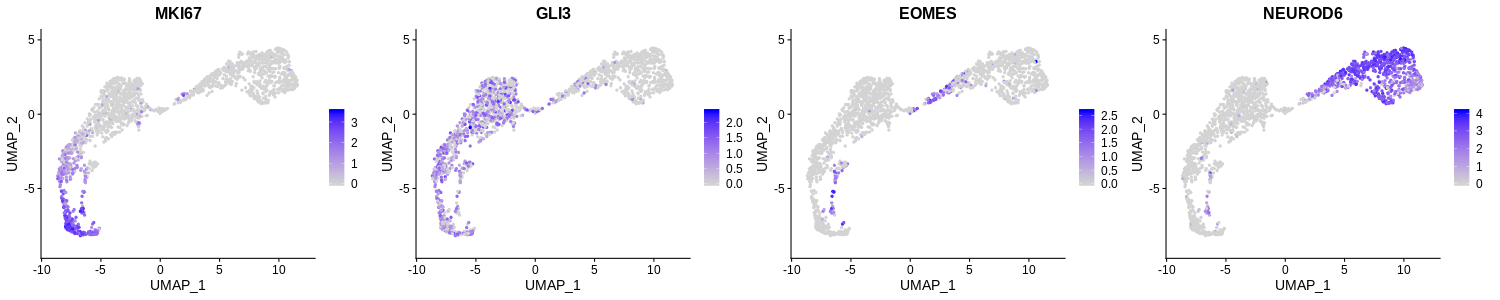
G2M 细胞不再处于分离的簇中,但它仍然混淆了细胞类型分化轨迹。例如,EOMES+ 细胞分别分布在两组中。我们需要进一步减少细胞周期效应。
正如上面简要提到的, ScaleData() 函数可以选择包含表示不需要的变化来源的变量。我们可以尝试利用它来进一步减少细胞周期的影响;但在此之前,我们需要为每个细胞生成与细胞周期相关的分数来描述它们的细胞周期状态。
seurat_dorsal <- CellCycleScoring(seurat_dorsal,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes,
set.ident = TRUE)
seurat_dorsal <- ScaleData(seurat_dorsal, vars.to.regress = c("S.Score", "G2M.Score"))
然后我们可以通过创建新的 UMAP 嵌入并绘制一些特征图来检查数据的外观
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
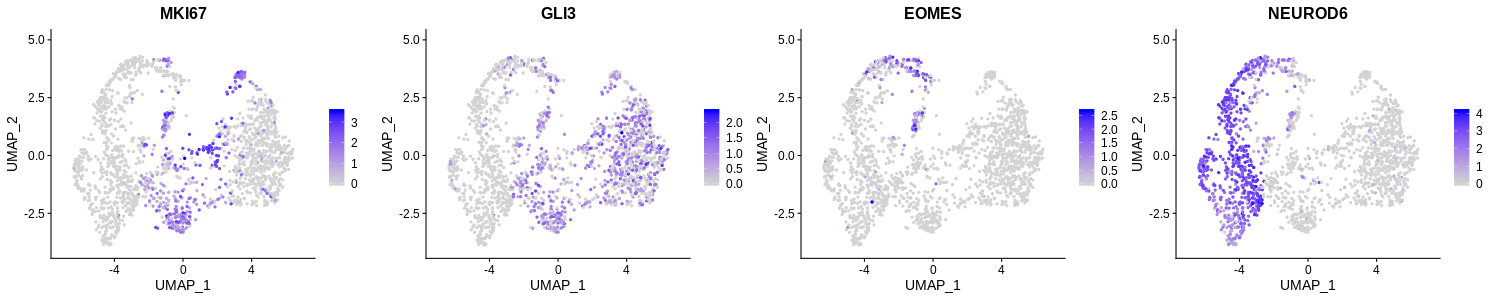
它并不完美,但至少我们不再看到两个独立的 EOMES+ 组。
现在让我们尝试运行扩散图来对细胞进行排序。
library(destiny)
dm <- DiffusionMap(Embeddings(seurat_dorsal, "pca")[,1:20])
dpt <- DPT(dm)
seurat_dorsal$dpt <- rank(dpt$dpt)
FeaturePlot(seurat_dorsal, c("dpt","GLI3","EOMES","NEUROD6"), ncol=4)
PS:这里估计的 dpt 的等级,而不是 dpt 本身,被用作最终的伪时间。两种选择都有优点和缺点。原则上,原始dpt不仅包含排序,还包含差异有多大。然而,其值范围通常由两侧的一些“异常值”主导,这些“异常值”很少被数据所代表。使用等级有助于在中等 DPT 时恢复这些变化。请随意尝试两者。
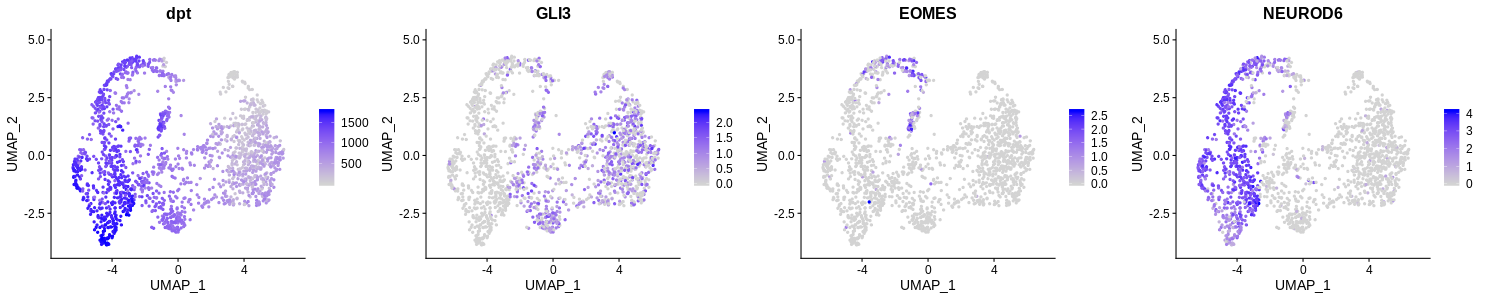
PS:与这里的示例不同,其中NPC的伪时间更短,也可能出现从神经元开始的伪时间序列。这是因为扩散伪时间,以及大多数其他基于相似性的伪时间分析方法,是没有方向性的。它估计了梯度,但如果你不告诉它,它不知道哪一端是源头。因此,如果你发现构建的伪时间走向了错误的方向,就翻转它(例如 seurat_dorsal$dpt <- rank(-dpt$dpt) )
为了可视化沿着构建的伪时间的表达变化,具有拟合曲线的散点图通常是一种直接的方法。
library(ggplot2)
plot1 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["GLI3",]),
xlab="Dpt", ylab="Expression", main="GLI3") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot2 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["EOMES",]),
xlab="Dpt", ylab="Expression", main="EOMES") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot3 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["NEUROD6",]),
xlab="Dpt", ylab="Expression", main="NEUROD6") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot1 + plot2 + plot3

11 保存结果¶
这些基本上是本教程第一部分中的所有内容,涵盖了可以对单个 scRNA-seq 数据集进行的大部分基本分析。在分析结束时,我们当然想要保存结果,可能是我们已经使用了一段时间的 Seurat 对象,这样下次我们就不需要再次重新运行所有分析。保存 Seurat 对象的方法与保存任何其他 R 对象相同。可以使用 saveRDS / readRDS 分别保存/加载每个 Seurat 对象,
saveRDS(seurat, file="DS1/seurat_obj_all.rds")
saveRDS(seurat_dorsal, file="DS1/seurat_obj_dorsal.rds")
seurat <- readRDS("DS1/seurat_obj_all.rds")
seurat_dorsal <- readRDS("DS1/seurat_obj_dorsal.rds")
save / load 将多个对象保存在一起 save(seurat, seurat_dorsal, file="DS1/seurat_objs.rdata")
load("DS1/seurat_objs.rdata")
主要代码汇总¶
library(Seurat)
# 设置多线程
library(future)
plan("multiprocess", workers = 4)
# 创建seurat对象
counts <- Read10X(data.dir = "data/DS1/")
seurat <- CreateSeuratObject(counts, project="DS1")
# 质控
print("质控")
seurat[["percent.mt"]] <- PercentageFeatureSet(seurat, pattern = "^MT[-\\.]")
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3, pt.size=0)
library(patchwork)
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
seurat <- subset(seurat, subset = nFeature_RNA > 500 & nFeature_RNA < 5000 & percent.mt < 5)
# 标准化
print("标准化")
seurat <- NormalizeData(seurat)
# 特征选择
print("特征选择")
seurat <- FindVariableFeatures(seurat, nfeatures = 3000)
top_features <- head(VariableFeatures(seurat), 20)
plot1 <- VariableFeaturePlot(seurat)
plot2 <- LabelPoints(plot = plot1, points = top_features, repel = TRUE)
plot1 + plot2
# 数据缩放
print("数据缩放")
seurat <- ScaleData(seurat)
# PCA分析
print("PCA分析")
seurat <- RunPCA(seurat, npcs = 50)
ElbowPlot(seurat, ndims = ncol(Embeddings(seurat, "pca")))
PCHeatmap(seurat, dims = 1:20, cells = 500, balanced = TRUE, ncol = 4)
# 非线性降维可视化
seurat <- RunTSNE(seurat, dims = 1:20)
seurat <- RunUMAP(seurat, dims = 1:20)
plot1 <- TSNEPlot(seurat)
plot2 <- UMAPPlot(seurat)
plot1 + plot2
plot1 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "tsne")
plot2 <- FeaturePlot(seurat, c("MKI67","NES","DCX","FOXG1","DLX2","EMX1","OTX2","LHX9","TFAP2A"),
ncol=3, reduction = "umap")
plot1 / plot2
# 聚类
print("聚类")
seurat <- FindNeighbors(seurat, dims = 1:20)
seurat <- FindClusters(seurat, resolution = 1)
plot1 <- DimPlot(seurat, reduction = "tsne", label = TRUE)
plot2 <- DimPlot(seurat, reduction = "umap", label = TRUE)
plot1 + plot2
# 注释细胞簇
ct_markers <- c("MKI67","NES","DCX","FOXG1", # G2M, NPC, neuron, telencephalon
"DLX2","DLX5","ISL1","SIX3","NKX2.1","SOX6","NR2F2", # ventral telencephalon related
"EMX1","PAX6","GLI3","EOMES","NEUROD6", # dorsal telencephalon related
"RSPO3","OTX2","LHX9","TFAP2A","RELN","HOXB2","HOXB5") # non-telencephalon related
DoHeatmap(seurat, features = ct_markers) + NoLegend()
cl_markers <- FindAllMarkers(seurat, only.pos = TRUE, min.pct = 0.25, logfc.threshold = log(1.2))
library(dplyr)
cl_markers %>% group_by(cluster) %>% top_n(n = 2, wt = avg_log2FC)
top10_cl_markers <- cl_markers %>% group_by(cluster) %>% top_n(n = 10, wt = avg_log2FC)
DoHeatmap(seurat, features = top10_cl_markers$gene) + NoLegend()
plot1 <- FeaturePlot(seurat, c("NEUROD2","NEUROD6"), ncol = 1)
plot2 <- VlnPlot(seurat, features = c("NEUROD2","NEUROD6"), pt.size = 0)
plot1 + plot2 + plot_layout(widths = c(1, 2))
# 拟时序分析
print("拟时序分析")
seurat_dorsal <- subset(seurat, subset = RNA_snn_res.1 %in% c(0,2,5,6,10))
seurat_dorsal <- FindVariableFeatures(seurat_dorsal, nfeatures = 2000)
VariableFeatures(seurat) <- setdiff(VariableFeatures(seurat), unlist(cc.genes))
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
seurat_dorsal <- CellCycleScoring(seurat_dorsal,
s.features = cc.genes$s.genes,
g2m.features = cc.genes$g2m.genes,
set.ident = TRUE)
seurat_dorsal <- ScaleData(seurat_dorsal, vars.to.regress = c("S.Score", "G2M.Score"))
seurat_dorsal <- RunPCA(seurat_dorsal) %>% RunUMAP(dims = 1:20)
FeaturePlot(seurat_dorsal, c("MKI67","GLI3","EOMES","NEUROD6"), ncol = 4)
library(destiny)
dm <- DiffusionMap(Embeddings(seurat_dorsal, "pca")[,1:20])
dpt <- DPT(dm)
seurat_dorsal$dpt <- rank(dpt$dpt)
FeaturePlot(seurat_dorsal, c("dpt","GLI3","EOMES","NEUROD6"), ncol=4)
library(ggplot2)
plot1 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["GLI3",]),
xlab="Dpt", ylab="Expression", main="GLI3") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot2 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["EOMES",]),
xlab="Dpt", ylab="Expression", main="EOMES") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot3 <- qplot(seurat_dorsal$dpt, as.numeric(seurat_dorsal@assays$RNA@data["NEUROD6",]),
xlab="Dpt", ylab="Expression", main="NEUROD6") +
geom_smooth(se = FALSE, method = "loess") + theme_bw()
plot1 + plot2 + plot3
第二部分:多样本分析¶
第三部分:带注释的数据集分析¶
第四部分:高级 scRNA-Seq 数据分析¶
参考¶
https://mp.weixin.qq.com/s/ArokNhoynciU5sPYaSSnMQ
Single-cell best practices-scanpy
单细胞转录组基础10讲 笔记 植物3篇单细胞文献 https://mp.weixin.qq.com/s/w3N5BkZWpdkEj5IfbxIFcQ
A single-cell RNA sequencing profiles the developmental landscape of Arabidopsis root
A single-cell analysis of the Arabidopsis vegetative shoot apex
Single-Cell Transcriptome Atlas and Chromatin Accessibility Landscape Reveal Differentiation Trajectories in the Rice Root
其它
单细胞测序数据报告看不懂?读这一篇就够了 https://mp.weixin.qq.com/s/o439heqFgCtuBFHbHy-ylw
本文阅读量 次本站总访问量 次