wgatools
本文档由魏文杰提供
wgatools: A cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
代码库:https://github.com/wjwei-handsome/wgatools
由来¶
全基因组比对,各个软件有各自的输出格式,如 MAF、SAM、PAF、delta 等,针对不同的下游操作和指定分析软件,有时需要格式转换,所以开发了此工具用于处理全基因组比对结果格式。
目前该工具已经开源在 github 上,部分 feature 也在开发中,欢迎大家使用 提建议 点赞。
安装¶
安装很简单,纯纯的 rust 写的,直接 cargo 安装就完事了,两种方式:
- 手动克隆 repo 编译
git clone https://github.com/wjwei-handsome/wgatools.git
cd wgatools
cargo build --release
./target/release/wgatools
- 直接指定 repo 安装
cargo install --git https://github.com/wjwei-handsome/wgatools.git
- 集群上调用
$ module load wgatools/0.1
使用¶
和市面上绝大多数的现代工具套件一样,采用 wgatools <subcommand> <options> 的形式进行调用,如下所示:
$ wgatools
wgatools -- a cross-platform and ultrafast toolkit for Whole Genome Alignment Files manipulation
Version: 0.1.0
Authors: Wenjie Wei <wjwei9908@gmail.com>
Usage: wgatools [OPTIONS] <COMMAND>
Commands:
maf2paf Convert MAF format to PAF format [aliases: m2p]
maf2chain Convert MAF format to Chain format [aliases: m2c]
paf2maf Convert PAF format to MAF format [aliases: p2m]
paf2chain Convert PAF format to Chain format [aliases: p2c]
chain2maf Convert Chain format to MAF format [aliases: c2m]
chain2paf Convert Chain format to PAF format [aliases: c2p]
maf-index Build index for MAF file [aliases: mi]
maf-ext Extract specific region from MAF file with index [aliases: me]
call Call Variants from MAF file [aliases: c]
tview View MAF file in terminal [aliases: tv]
stat Statistics for Alignment file [aliases: st]
dotplot TEST: Plot dotplot for Alignment file [aliases: dp]
filter Filter records for Alignment file [aliases: fl]
rename Rename MAF records with prefix [aliases: rn]
maf2sam TEST: maf2sam [aliases: m2s]
pafcov TEST: pafcov [aliases: pc]
pafpseudo TEST: generate pesudo maf from paf [aliases: pp]
trimovp TEST: trim overlap for paf [aliases: tr]
help Print this message or the help of the given subcommand(s)
Options:
-h, --help Print help (see more with '--help')
-V, --version Print version
GLOBAL:
-o, --outfile <OUTFILE> Output file ("-" for stdout) [default: -]
-r, --rewrite Bool, if rewrite output file [default: false]
-t, --threads <THREADS> Threads, default 1 [default: 1]
-v, --verbose... Logging level [-v: Info, -vv: Debug, -vvv: Trace, defalut: Warn]
其中有些参数是全局的,这些在所有子命令中都是通用的,比如指定输出的文件,指定是否覆盖已有的文件,指定线程数,指定日志等级。
同时提供了 alias 的重命名,可以不用输入完整的命令,比如 maf2paf 可以简写为 m2p, paf2maf 可以简写为 p2m 等等。
示例¶
格式的互相转换¶
这个是本工具的核心功能,用于三种格式之间的互相转换。用法简单,以 maf2paf 为例:
$ wgatools maf2paf test.maf > test.paf
需要将不包含序列信息的 PAF 格式转成包含序列 MAF 格式,需要指定基因组文件,支持gz压缩格式,若无索引会构建索引。示例如下:
$ wgatools paf2maf test.paf -g target.fa -q query.fa > test.maf
支持标准输入输出,所有可以套娃:
$ cat test.maf | wgatools maf2paf | wgatools paf2maf -g target.fa -q query.fa > test.maf
$ wgatools paf2chain test.paf | wgatools chain2maf -g target.fa -q query.fa | wgatools maf2chain | wgatools chain2paf > funny.paf
MAF 的索引和区间提取¶
其实 MAF 是包含了全基因组比对最原始和最全面的信息,在看 MAF 文件,有时候会有需求想看指定区间的比对情况,那么 MAF 文件一行又非常长,而且没有坐标的提示,很难找到感兴趣的区间。该子命令可以快速提取指定染色体和区间的比对信息,于是就开发了 MAF 的索引和提取功能。示例如下:
$ wgatools maf-index test.maf
该功能无需指定输出文件,也不会输出到标准输出,会自动产生后缀为 .index 的索引文件,展示设计为了json格式,因为这个包含的数据其实不多,没必要用上protobuf或者bincode之类的二进制格式。
有了索引之后就可以提取区间,支持两种方式的区间:
chr:start-end形式的区间
这里采用了正则表达式方案来匹配输入,所以start和end如果是非数字会报错。同时会检查start是否小于end.
可以用逗号分隔多个区间,比如:
$ wgatools maf-extract test.maf -r chr1:1-10,chr2:66-888,chr3:100-50,chr_no:1-10,x:y-z
例如这个例子中的chr3:100-50 , chr_no:1-10,x:y-z都会报错:
ERROR Parse Genome Region Error By: Region `x:y-z` is match the format of `chr:start-end`
ERROR Parse Genome Region Error By: Start `100` is larger than end `50`
bed格式文件的输入
可以在文件中指定多个区间,每行一个,格式为chr\tstart\tend,比如:
$ wgatools maf-extract test.mfa -f region.bed
这两种区间输入方式,可以同时指定,会合并在一起。
如果输入的区间有一个格式错了,就会退出程序。
如果输入的区间没有匹配,会警告未匹配的区间,并且跳过。
统计¶
支持 MAF/PAF 格式,可以支持是否合并同一染色体,如下:
$ wgatools stat test.maf
ref_name ref_size ref_start query_name query_size query_start aligned_size unaligned_size identity similarity matched mismatched ins_event del_event ins_size del_size inv_event inv_size inv_ins_event inv_ins_size inv_del_event inv_del_size
XXX.chr2 243675191 232285008 YYY.chr2 241186694 229666595 11007552 232667639 0.47867247 0.4921803 5269012 148688 11831 11746 5229899 5315762 1 667431.0 1115 546710 1089 274090
XXX.chr6 181357234 173309259 YYY.chr6 178141476 169794562 7338167 174019067 0.5532852 0.5652489 4060099 87792 8540 8825 3361418 3190276 0 0.0 0 0 0 0
XXX.chr8 182411202 142613895 YYY.chr8 184335743 142573702 2447590 179963612 0.42162004 0.43657106 1031953 36594 0 0 0 0 1 2241621.5 2575 967106 2532 1379043
XXX.chr9 163004744 162104206 YYY.chr9 166955280 166049855 792919 162211825 0.64267725 0.652846 509591 8063 489 484 301376 275265 0 0.0 0 0 0 0
$ wgatools stat test.maf -e
ref_name ref_size ref_start query_name query_size query_start aligned_size unaligned_size identity similarity matched mismatched ins_event del_event ins_size del_size inv_event inv_size inv_ins_event inv_ins_size inv_del_event inv_del_size
XXX.chr2 243675191 239959575 YYY.chr2 241186694 237977650 531121 0 0.45960337 0.48394057 244105 12926 0 0 0 0 1 667431.0 1115 546710 1089 274090
XXX.chr2 243675191 240631599 YYY.chr2 241186694 238865793 2853246 0 0.4797739 0.48980528 1368913 28622 2692 2662 718206 1455711 0 0.0 0 0 0 0
XXX.chr2 243675191 232285008 YYY.chr2 241186694 229666595 7623185 0 0.47958878 0.49364328 3655994 107140 9139 9084 4511693 3860051 0 0.0 0 0 0 0
XXX.chr6 181357234 173309259 YYY.chr6 178141476 169794562 7338167 0 0.5532852 0.5652489 4060099 87792 8540 8825 3361418 3190276 0 0.0 0 0 0 0
XXX.chr8 182411202 142613895 YYY.chr8 184335743 142573702 2447590 0 0.42162004 0.43657106 1031953 36594 0 0 0 0 1 2241621.5 2575 967106 2532 1379043
XXX.chr9 163004744 162104206 YYY.chr9 166955280 166049855 792919 0 0.64267725 0.652846 509591 8063 489 484 301376 275265 0 0.0 0 0 0 0
过滤¶
在某些比对中,我们会有contig和chromosome比对到一起的情况,通常我们并不需要这些比对,所以可以过滤掉。
$ wgatools filter test.maf -q 1000000 > filt.maf
在该子命令中,针对wfmash 比对软件产生的all-to-all 比对的 PAF 文件,可以过滤最低比对区域大小的target-query对,比如跨染色体的比对,如下:
$ wgatools filter -a 1000000 -f paf wfmash_out.paf > wfmash_out_filter1M.paf
该示例中,-f参数用于指定格式为PAF,-a为最小比对区域大小
鉴定变异¶
MAF 格式完整地记录了每个碱基的比对情况,所以可以用来鉴定变异,支持SNP/INS/DEL/INV这几种显式的变异类型。默认参数不输出SNP和长度小于 50 的INS和DEL,示例如下:
$ wgatools call -s sample_name test.maf > call.vcf
本功能只支持鉴定显式的变异,如果要鉴定
DUP等其他染色体重排变异,还需要提取上下游序列来比对预测,也许之后可以加上。
产生的结果中,REF和ALT区域为产生变异的序列信息;INFO区域包含结构变异的种类、长度、终止位点等;FORMAT区域中包含基因型和该变异在query基因组上的位置信息(如插入了 query.chr1:6000-6060 区域的序列)。
终端可视化¶
之前提到, 查看 MAF 文件非常麻烦,而且就算提取了指定区间,也没有坐标的指示,而且换一下 target 和 query 的上下位置也很麻烦,所以就直接开发了一个终端可视化的功能,可以在终端中查看 MAF 文件,使用方法如下:
$ wgatools tview test.maf
会出现一个终端可视化页面,如下:

进入该页面后,按下 ◄► 可以左右滑动,在输入参数中可以指定滑动步长,默认为 10,按下 q 可以退出。
按下 g 可以调出导航窗口,其中左侧为可选的序列名称,右侧为选中序列的可选区间,可以按下 Tab切换左右选择窗口,可以按▲▼来选择序列和区间,同时下方的输入框会随之更新,也可以回车删除和键盘输入来指定区间,检查输入是合法区间后,可以按下 Enter 可以跳转到指定的序列和区间,同时会退出导航窗口,也可以按下Esc来退出导航窗口。
重命名¶
在用 AnchorWave、minimap2、wfmash 等软件进行比对后,有时候由于染色体名称命名的一致性,导致比对结果中 target 和 query 的染色体名称相同,比如都为chr1,难以区分且不利于下游分析,对于 PAF 格式修改相对容易,但是 MAF 较麻烦,所以该子命令可以快速添加前缀,例如:
$ wgatools rename --prefixs Zm-B73.,Zm-SK. input.maf > rename.maf
这样输出文件的染色体名称就会变成Zm-B73.chr1和Zm-SK.chr1
基因组比对覆盖度计算 (试验性)¶
在比较基因组学分析中,我们经常会将多个基因组比对到最常用的参考基因组上,在获得全基因组比对结果后,可以将多个基因组比对结果的 PAF 格式(如果是 MAF 格式,可以用maf2paf转换)合并到一个all-to-all的 PAF 结果中,当然最方便的是使用wfmash直接获得,然后用该子命令计算参考基因组上的比对覆盖度,如下所示:
$ wgatools pafcov all.paf > all.cov.bed
产生的结果为bed格式,其中每一行表示为基因组上每个位置上的覆盖度,该结果可以用于下游的保守位点分析等。
伪 MAF 的生成 (试验性)¶
和上一个例子类似,获得了all-to-all的 PAF 结果后,可以将其转换为该种伪MAF格式,之所以称之为伪,因为是以 target 基因组为基准,所以该 MAF 中不包含 query 基因组的插入序列信息,但是由于每个基因组都可以作为 target,所以最终转换的结果也包含了无损的全量信息,示例如下:
$ wgatools pafpseudo -f all.fa.gz all.paf -o out_dir -t 10
该示例中,参数-f用于指定基因组序列文件,支持gz压缩格式。参数-o不允许标准输出,而指定为目录,该输出目录中会包含所有转换得到的MAF。参数-t表示线程树,若资源条件允许,将其设置为 PAF 文件中target染色体的数量最佳(即cut -f6 all.paf|sort -u|wc -l )。结果如下图所示:

该结果可以用于下游系统发育分析等。
使用体验¶
速度¶
Rust 语言加持,软件整体速度较快
比较¶
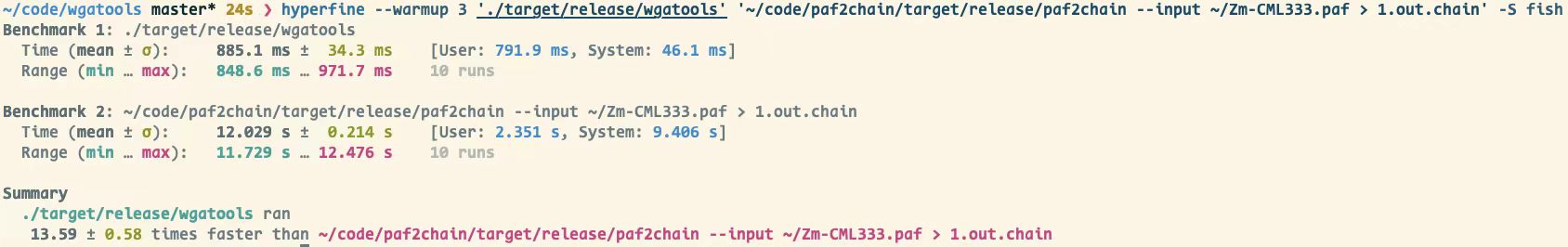
由于其中系列功能在市面上大多数是个人脚本,暂未进行比较,但是相比同样是 Rust 编写的paf2chain,使用hypferfine进行合理评估后,本工具速度大约可以提升 14 倍。
内存¶
构建本工具时,对于不同文件的读取设计了合理的迭代器,内存开销较低。
注意:在使用call子命令时,当时未进行优化,可能内存要求较高。后续会改进。
错误处理¶
本工具定义了较为全面完整的错误类型和传递系统,若发生报错,可以通过报错信息获得帮助。
TODO¶
该工具从第一个子功能开始,历经约五个月,仍处于开发状态,目前已确定的处于开发计划中的新功能和 feature 有:
- Rust Test cases
- API Document build
- MAF chunk
- MAF/PAF 的 dotplot 出图
- ...
贡献¶
ideas:
🙋🏻欢迎贡献 issue、PR!
本文阅读量 次本站总访问量 次